

New mechanisms of pulmonary arterial hypertension: role of Ca²⁺ signaling
- PMID: 22245772
- PMCID: PMC3330808
- DOI: 10.1152/ajpheart.00944.2011
New mechanisms of pulmonary arterial hypertension: role of Ca²⁺ signaling
Abstract
Pulmonary arterial hypertension (PAH) is a severe and progressive disease that usually culminates in right heart failure and death if left untreated. Although there have been substantial improvements in our understanding and significant advances in the management of this disease, there is a grim prognosis for patients in the advanced stages of PAH. A major cause of PAH is increased pulmonary vascular resistance, which results from sustained vasoconstriction, excessive pulmonary vascular remodeling, in situ thrombosis, and increased pulmonary vascular stiffness. In addition to other signal transduction pathways, Ca(2+) signaling in pulmonary artery smooth muscle cells (PASMCs) plays a central role in the development and progression of PAH because of its involvement in both vasoconstriction, through its pivotal effect of PASMC contraction, and vascular remodeling, through its stimulatory effect on PASMC proliferation. Altered expression, function, and regulation of ion channels and transporters in PASMCs contribute to an increased cytosolic Ca(2+) concentration and enhanced Ca(2+) signaling in patients with PAH. This review will focus on the potential pathogenic role of Ca(2+) mobilization, regulation, and signaling in the development and progression of PAH.
Figures
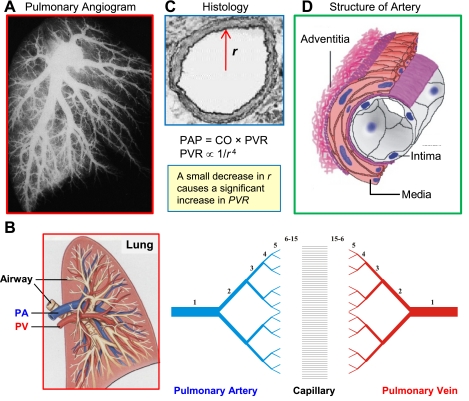




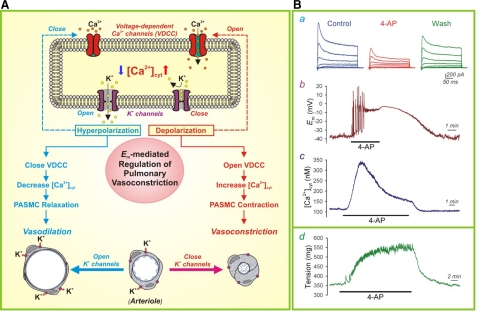
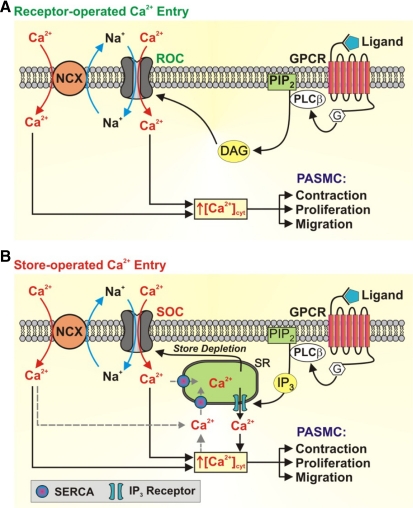




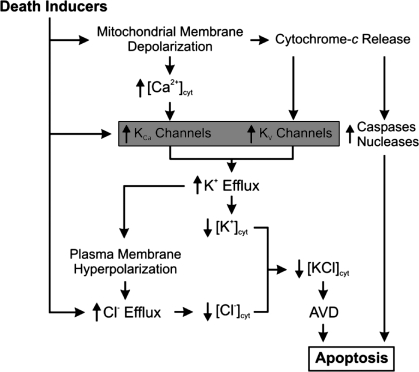

References
-
- Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress”. Circulation 102: 2781–2791, 2000 - PubMed
-
- Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv21, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101: 2319–2330, 1998 - PMC - PubMed
-
- Archer SL, Wu XC, Thebaud B, Nsair A, Bonnet S, Tyrrell B, McMurtry MS, Hashimoto K, Harry G, Michelakis ED. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ Res 95: 308–318, 2004 - PubMed
-
- Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science 253: 551–555, 1991 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous