

Pathophysiology of pulmonary hypertension in acute lung injury
- PMID: 22246001
- PMCID: PMC3362157
- DOI: 10.1152/ajplung.00355.2011
Pathophysiology of pulmonary hypertension in acute lung injury
Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome are characterized by protein rich alveolar edema, reduced lung compliance, and acute severe hypoxemia. A degree of pulmonary hypertension (PH) is also characteristic, higher levels of which are associated with increased morbidity and mortality. The increase in right ventricular (RV) afterload causes RV dysfunction and failure in some patients, with associated adverse effects on oxygen delivery. Although the introduction of lung protective ventilation strategies has probably reduced the severity of PH in ALI, a recent invasive hemodynamic analysis suggests that even in the modern era, its presence remains clinically important. We therefore sought to summarize current knowledge of the pathophysiology of PH in ALI.
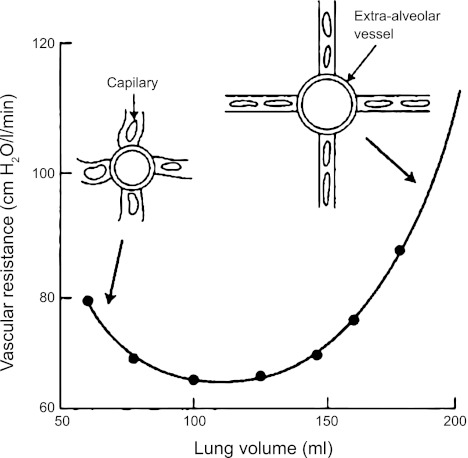
Figures




References
-
- Afshari A, Brok J, Moller AM, Wetterslev J. Aerosolized prostacyclin for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Cochrane Database Syst Rev 8: CD007733, 2010 - PubMed
-
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 4: 499–511, 2004 - PubMed
-
- Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 2: 319–323, 1967
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases