

Robust rank aggregation for gene list integration and meta-analysis
- PMID: 22247279
- PMCID: PMC3278763
- DOI: 10.1093/bioinformatics/btr709
Robust rank aggregation for gene list integration and meta-analysis
Abstract
Motivation: The continued progress in developing technological platforms, availability of many published experimental datasets, as well as different statistical methods to analyze those data have allowed approaching the same research question using various methods simultaneously. To get the best out of all these alternatives, we need to integrate their results in an unbiased manner. Prioritized gene lists are a common result presentation method in genomic data analysis applications. Thus, the rank aggregation methods can become a useful and general solution for the integration task.
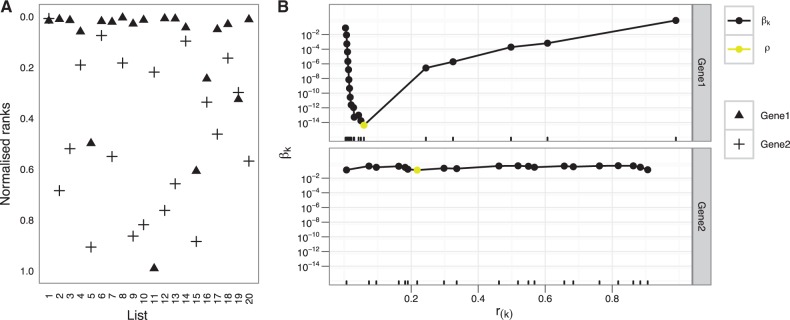
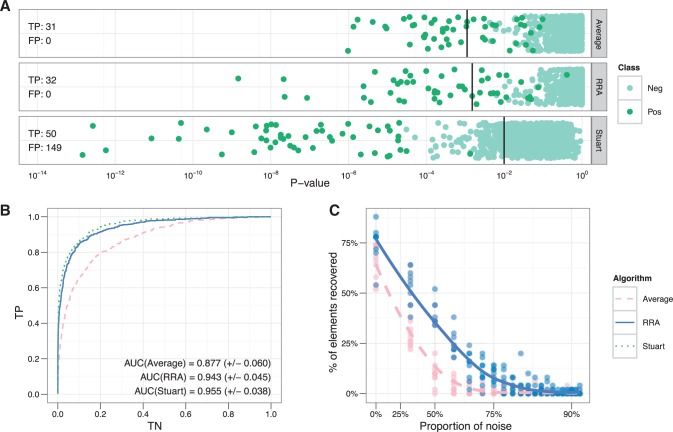
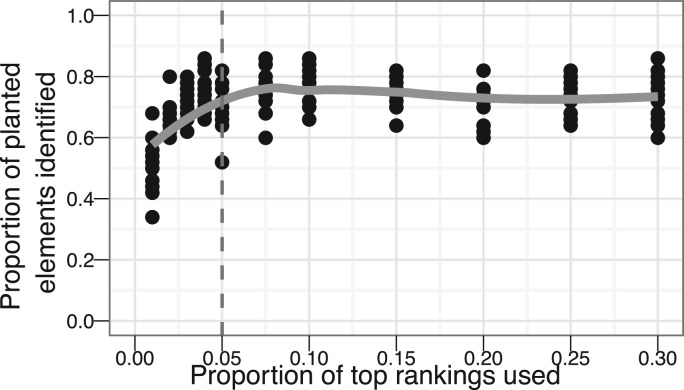
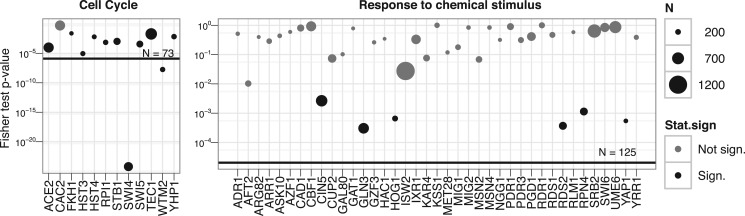
Results: Standard rank aggregation methods are often ill-suited for biological settings where the gene lists are inherently noisy. As a remedy, we propose a novel robust rank aggregation (RRA) method. Our method detects genes that are ranked consistently better than expected under null hypothesis of uncorrelated inputs and assigns a significance score for each gene. The underlying probabilistic model makes the algorithm parameter free and robust to outliers, noise and errors. Significance scores also provide a rigorous way to keep only the statistically relevant genes in the final list. These properties make our approach robust and compelling for many settings.
Availability: All the methods are implemented as a GNU R package RobustRankAggreg, freely available at the Comprehensive R Archive Network http://cran.r-project.org/.
Figures
References
-
- Aerts S., et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. - PubMed
-
- Bie T.D., et al. Kernel-based data fusion for gene prioritization. Bioinformatics. 2007;23:i125–i132. - PubMed
-
- Boulesteix A., Slawski M. Stability and aggregation of ranked gene lists. Brief. Bioinformatics. 2009;10:556. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
