

Human genome sequencing in health and disease
- PMID: 22248320
- PMCID: PMC3656720
- DOI: 10.1146/annurev-med-051010-162644
Human genome sequencing in health and disease
Abstract
Following the "finished," euchromatic, haploid human reference genome sequence, the rapid development of novel, faster, and cheaper sequencing technologies is making possible the era of personalized human genomics. Personal diploid human genome sequences have been generated, and each has contributed to our better understanding of variation in the human genome. We have consequently begun to appreciate the vastness of individual genetic variation from single nucleotide to structural variants. Translation of genome-scale variation into medically useful information is, however, in its infancy. This review summarizes the initial steps undertaken in clinical implementation of personal genome information, and describes the application of whole-genome and exome sequencing to identify the cause of genetic diseases and to suggest adjuvant therapies. Better analysis tools and a deeper understanding of the biology of our genome are necessary in order to decipher, interpret, and optimize clinical utility of what the variation in the human genome can teach us. Personal genome sequencing may eventually become an instrument of common medical practice, providing information that assists in the formulation of a differential diagnosis. We outline herein some of the remaining challenges.
Figures
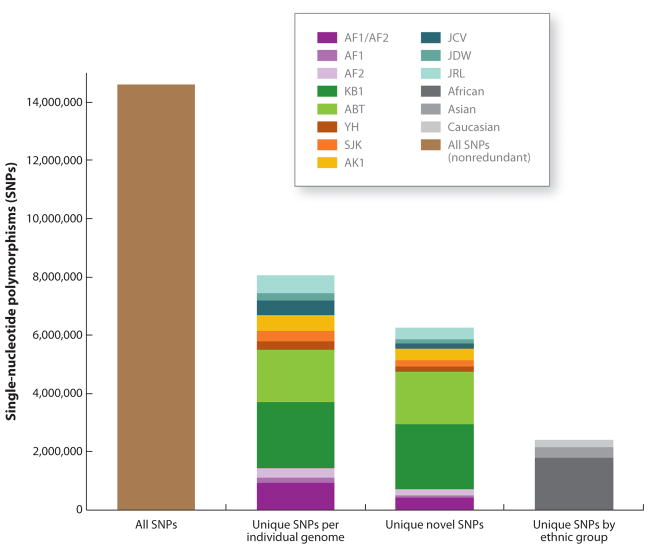
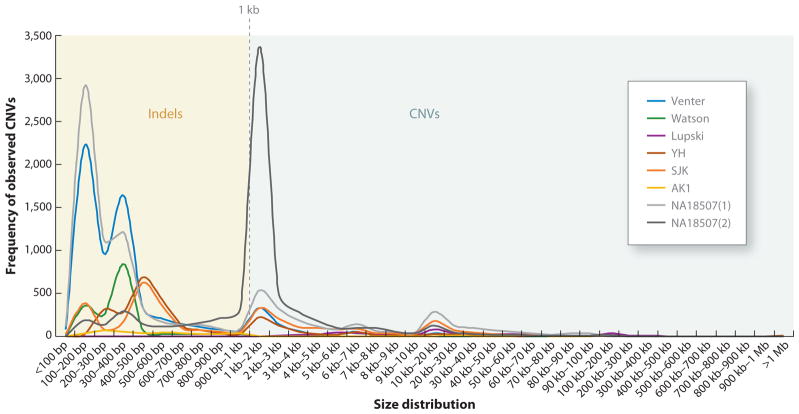
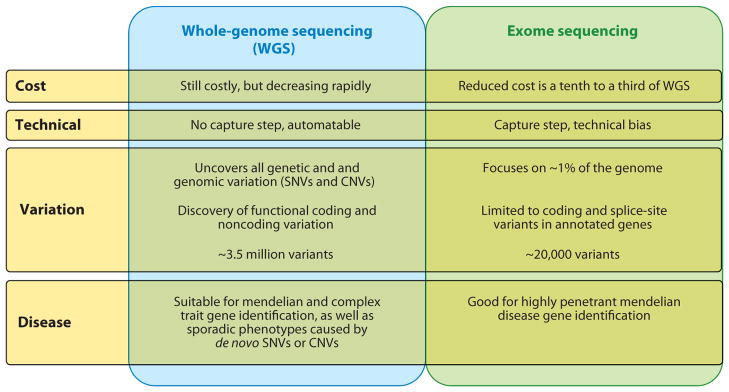
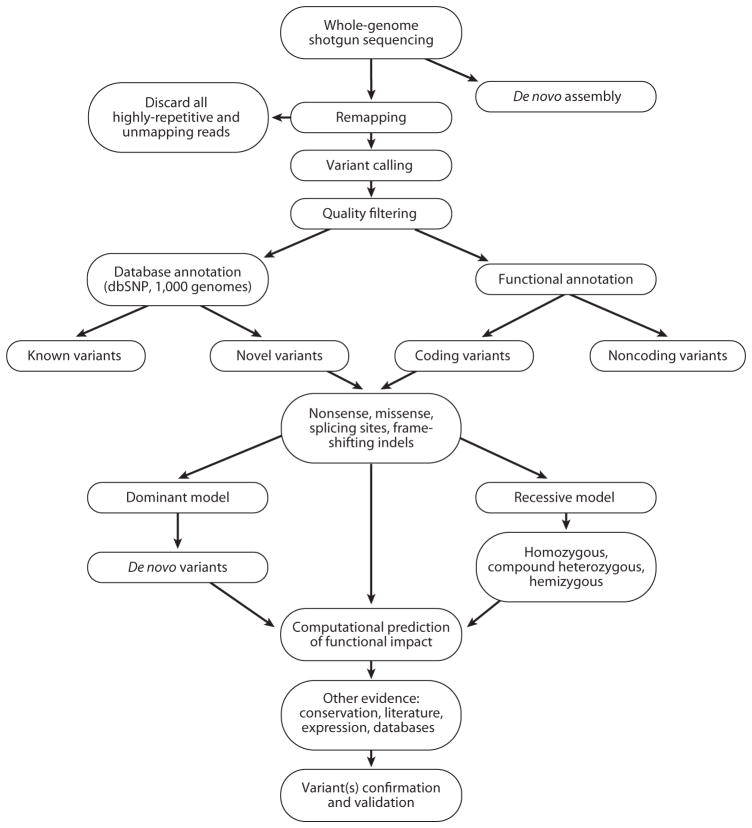
References
-
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. - PubMed
-
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. - PubMed
-
- The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–96. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
