

Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice
- PMID: 22249462
- PMCID: PMC3432922
- DOI: 10.1097/NEN.0b013e318244b635
Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice
Abstract
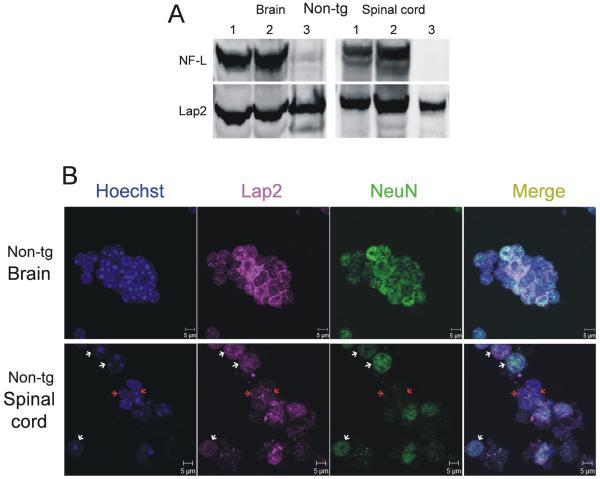
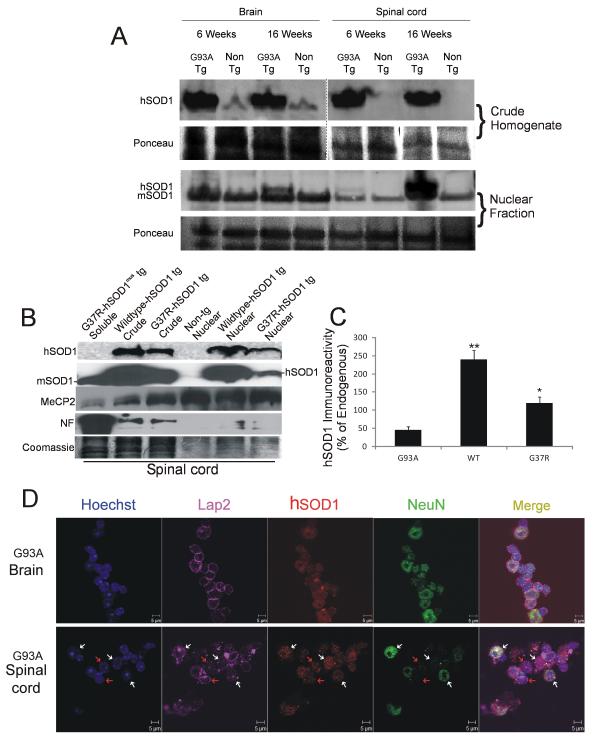
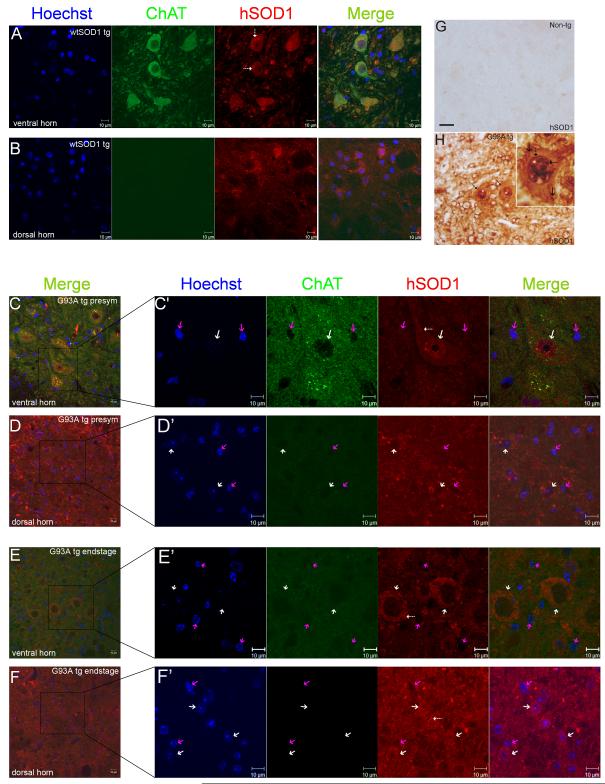
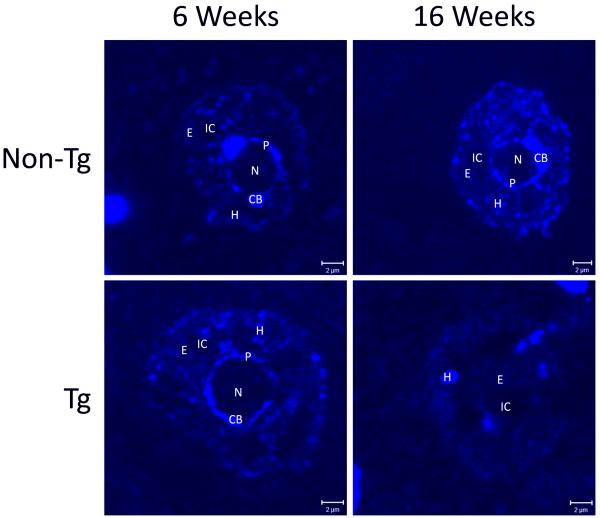
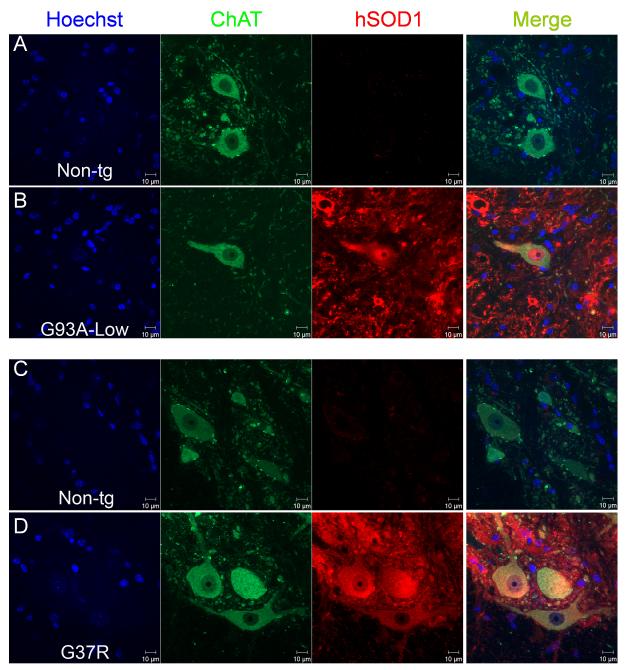
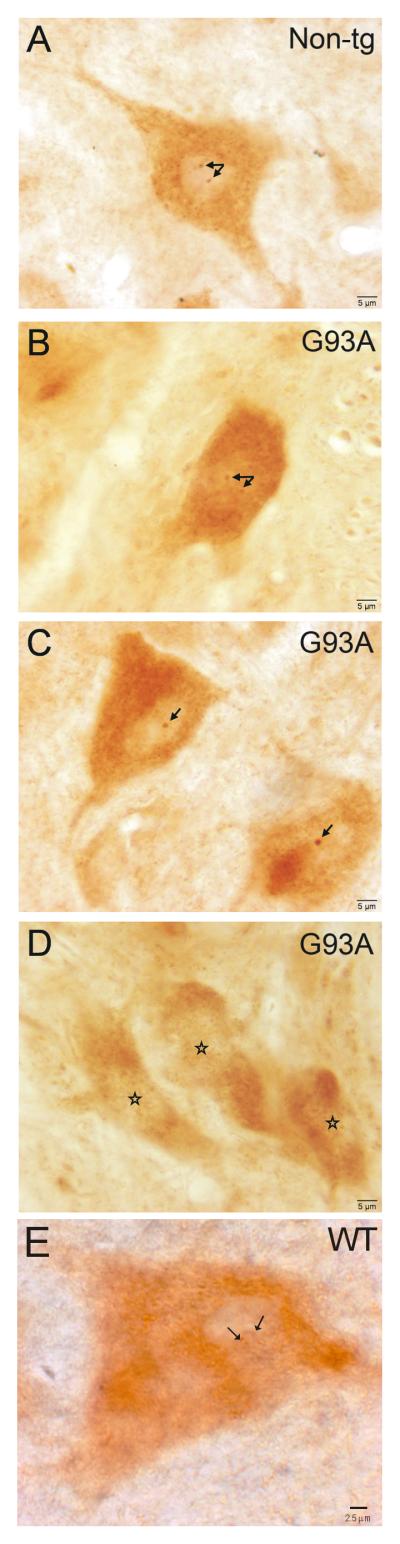
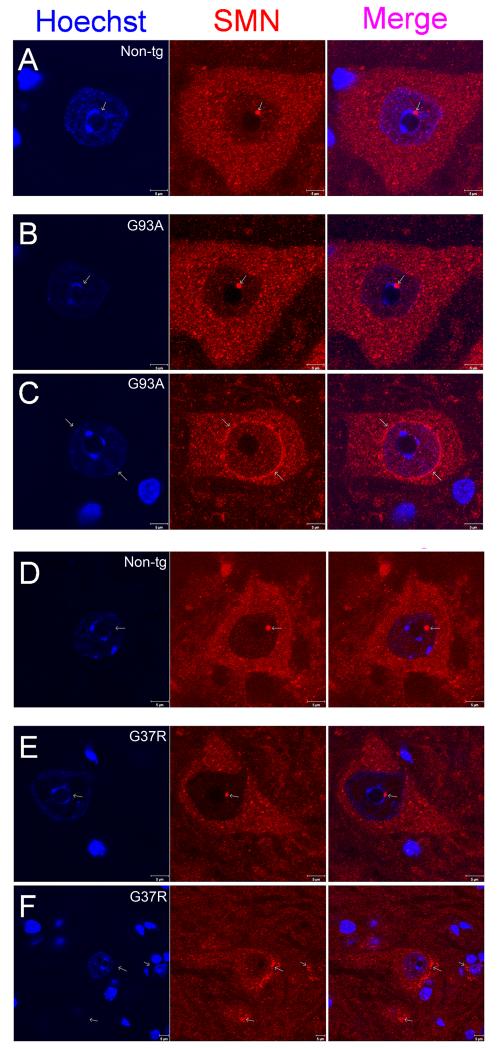
Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset neurodegenerative disease that causes degeneration of motor neurons and paralysis. Approximately 20% of familial ALS cases have been linked to mutations in the copper/zinc superoxide dismutase (SOD1) gene, but it is unclear how mutations in the protein result in motor neuron degeneration. Transgenic (tg) mice expressing mutated forms of human SOD1 (hSOD1) develop clinical and pathological features similar to those of ALS. We used tg mice expressing hSOD1-G93A, hSOD1-G37R, and hSOD1-wild-type to investigate a new subcellular pathology involving mutant hSOD1 protein prominently localizing to the nuclear compartment and disruption of the architecture of nuclear gems. We developed methods for extracting relatively pure cell nucleus fractions from mouse CNS tissues and demonstrate a low nuclear presence of endogenous SOD1 in mouse brain and spinal cord, but prominent nuclear accumulation of hSOD1-G93A, -G37R, and -wild-type in tg mice. The hSOD1 concentrated in the nuclei of spinal cord cells, particularly motor neurons, at a young age. The survival motor neuron protein (SMN) complex is disrupted in motor neuron nuclei before disease onset in hSOD1-G93A and -G37R mice; age-matched hSOD1-wild-type mice did not show SMN disruption despite a nuclear presence. Our data suggest new mechanisms involving hSOD1 accumulation in the cell nucleus and mutant hSOD1-specific perturbations in SMN localization with disruption of the nuclear SMN complex in ALS mice and suggest an overlap of pathogenic mechanisms with spinal muscular atrophy.
Figures
References
-
- Chou SM. Pathology-light microscopy of amyotrophic lateral sclerosis. In: Smith RA, editor. Handbook of Amyotrophic Lateral Sclerosis. Marcel Deckker, Inc.; New York, NY: 1992. pp. 133–81.
-
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. - PubMed
-
- Bendotti C, Carrì MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004;10:393–400. 2004. - PubMed
-
- Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4. - PubMed
-
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
