

Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease
- PMID: 22251701
- PMCID: PMC3266791
- DOI: 10.1172/JCI60070
Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease
Abstract
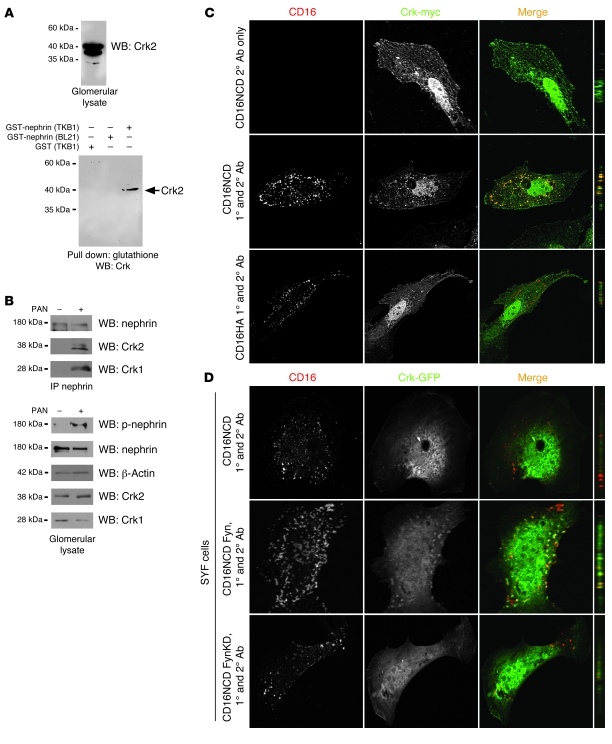
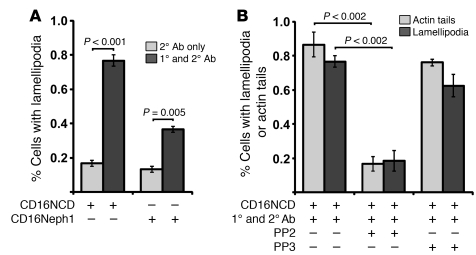
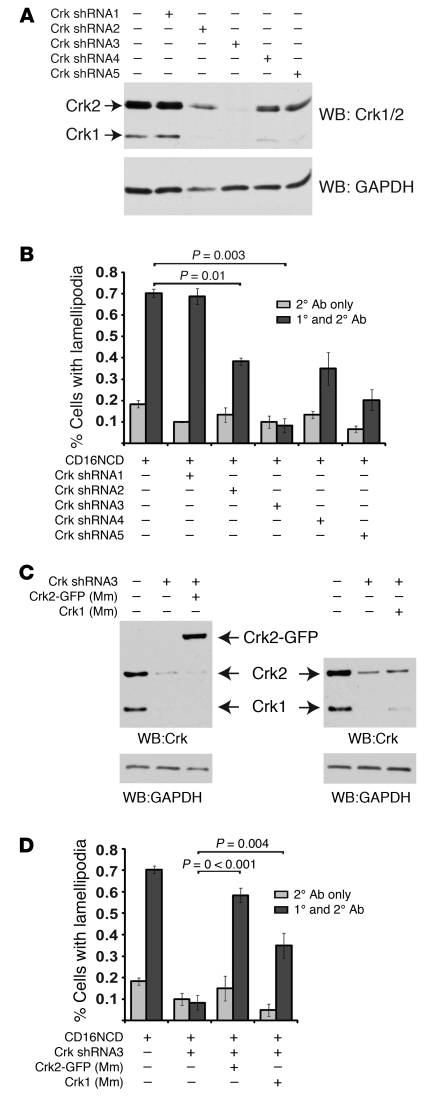
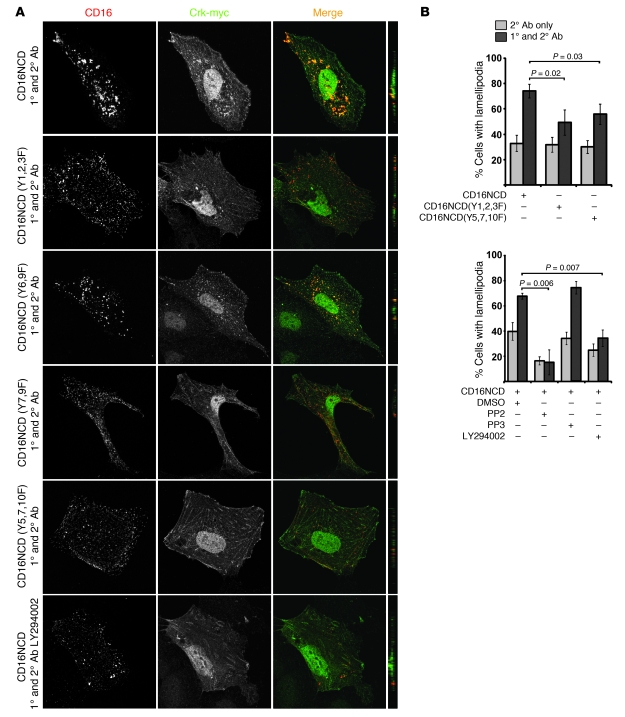
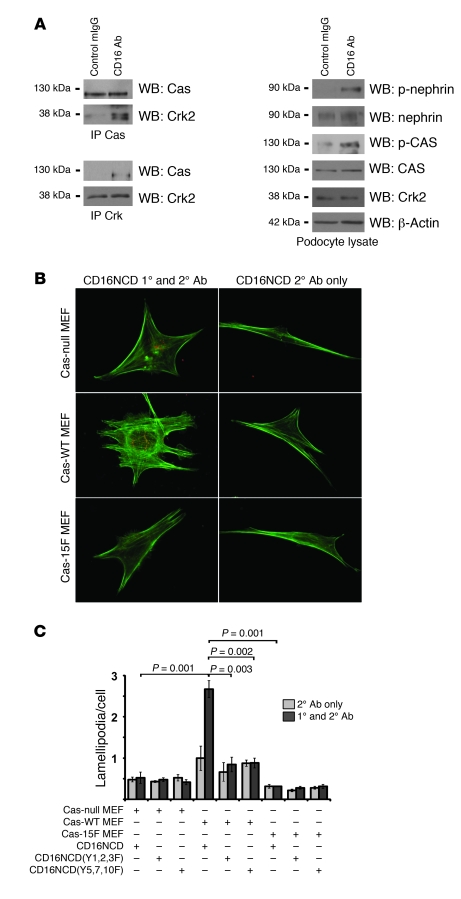
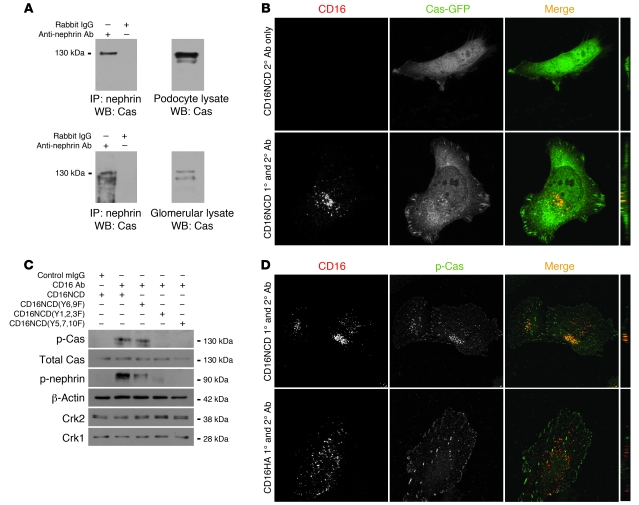
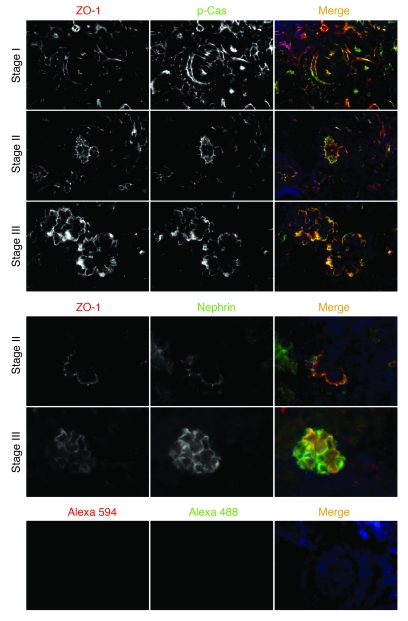
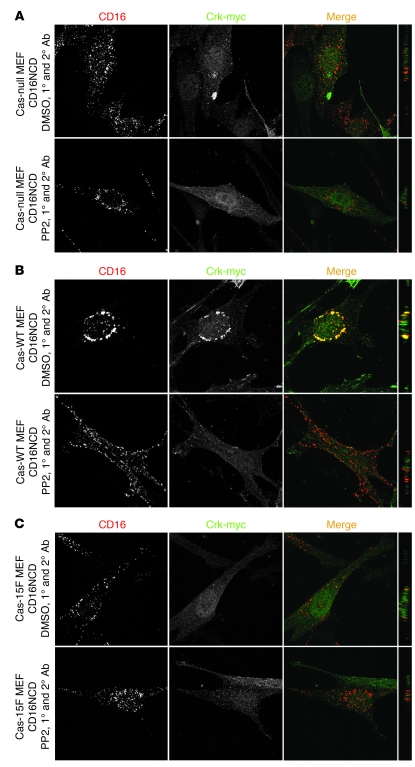
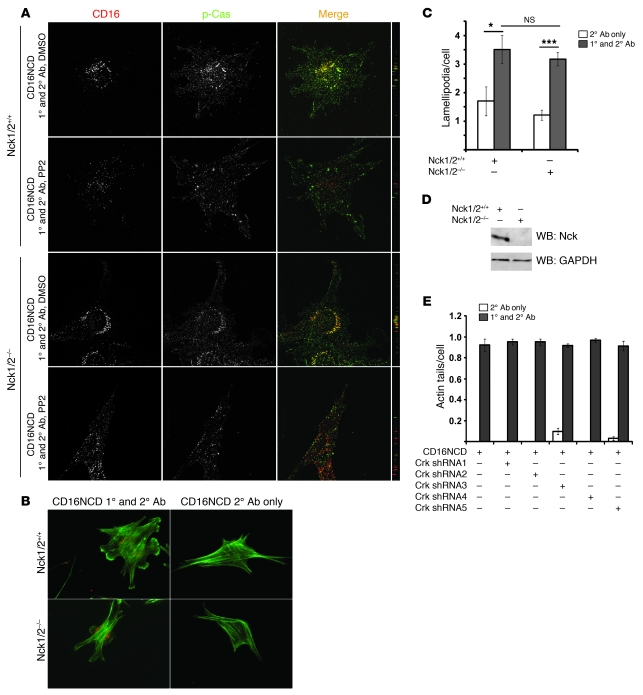
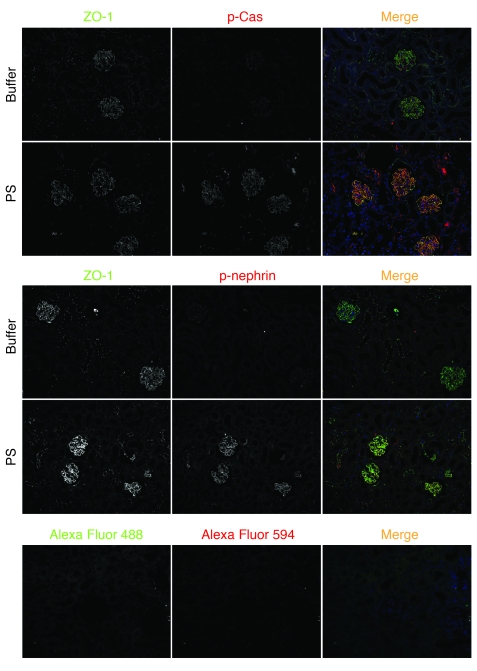
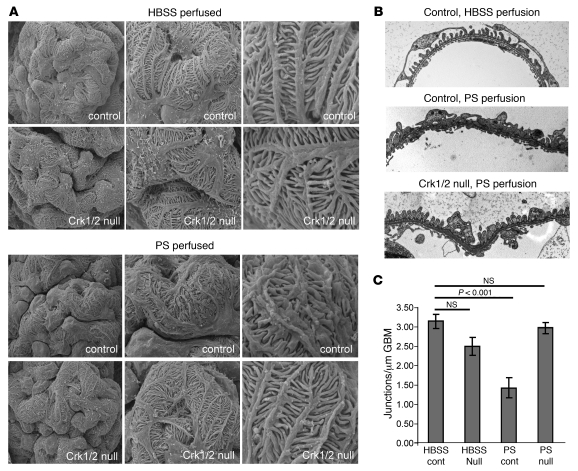
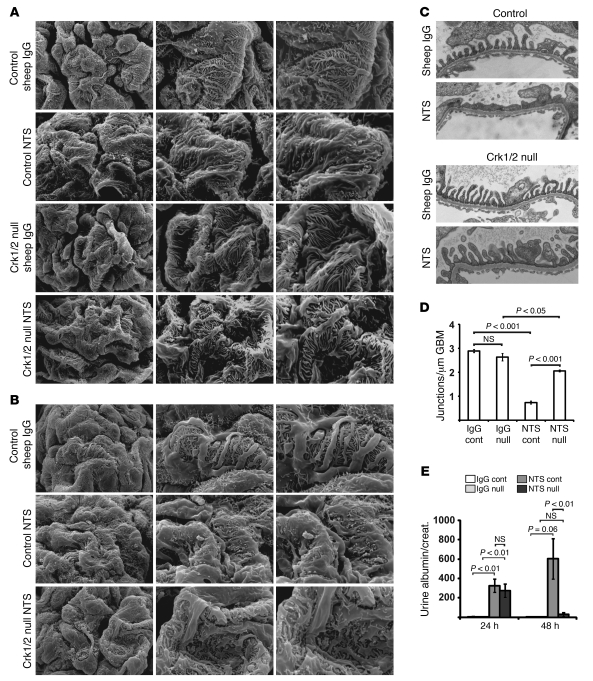
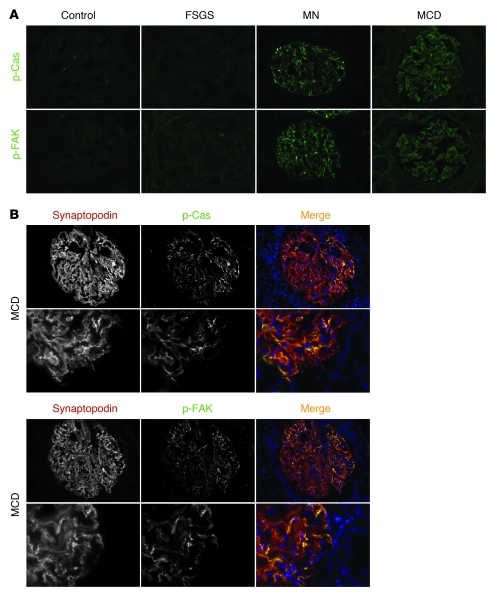
The morphology of healthy podocyte foot processes is necessary for maintaining the characteristics of the kidney filtration barrier. In most forms of glomerular disease, abnormal filter barrier function results when podocytes undergo foot process spreading and retraction by remodeling their cytoskeletal architecture and intercellular junctions during a process known as effacement. The cell adhesion protein nephrin is necessary for establishing the morphology of the kidney podocyte in development by transducing from the specialized podocyte intercellular junction phosphorylation-mediated signals that regulate cytoskeletal dynamics. The present studies extend our understanding of nephrin function by showing that nephrin activation in cultured podocytes induced actin dynamics necessary for lamellipodial protrusion. This process required a PI3K-, Cas-, and Crk1/2-dependent signaling mechanism distinct from the previously described nephrin-Nck1/2 pathway necessary for assembly and polymerization of actin filaments. Our present findings also support the hypothesis that mechanisms governing lamellipodial protrusion in culture are similar to those used in vivo during foot process effacement in a subset of glomerular diseases. In mice, podocyte-specific deletion of Crk1/2 prevented foot process effacement in one model of podocyte injury and attenuated foot process effacement and associated proteinuria in a delayed fashion in a second model. In humans, focal adhesion kinase and Cas phosphorylation - markers of focal adhesion complex-mediated Crk-dependent signaling - was induced in minimal change disease and membranous nephropathy, but not focal segmental glomerulosclerosis. Together, these observations suggest that activation of a Cas-Crk1/2-dependent complex is necessary for foot process effacement observed in distinct subsets of human glomerular diseases.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
