

Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells
- PMID: 22251703
- PMCID: PMC3266798
- DOI: 10.1172/JCI61034
Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells
Abstract
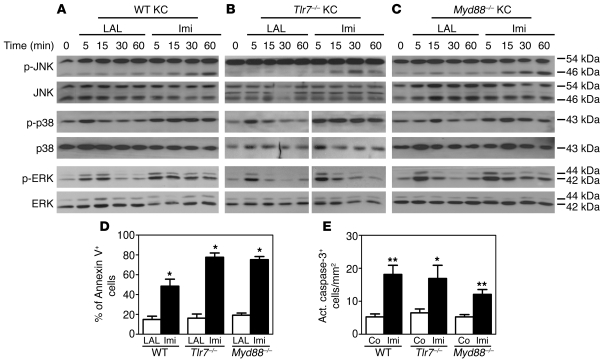
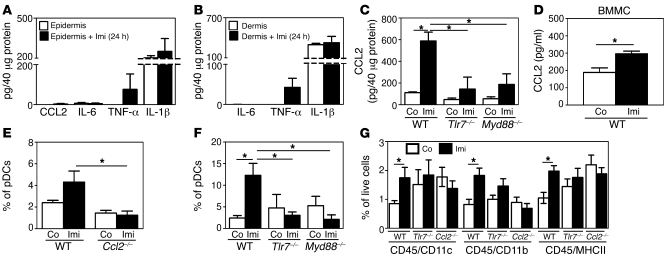
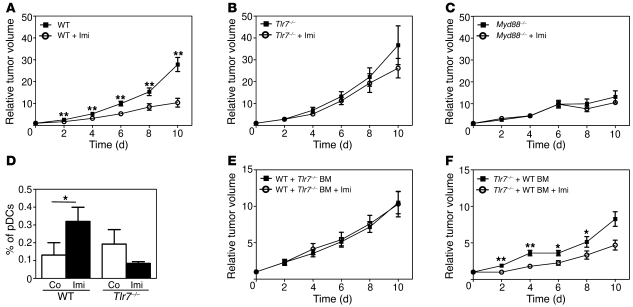
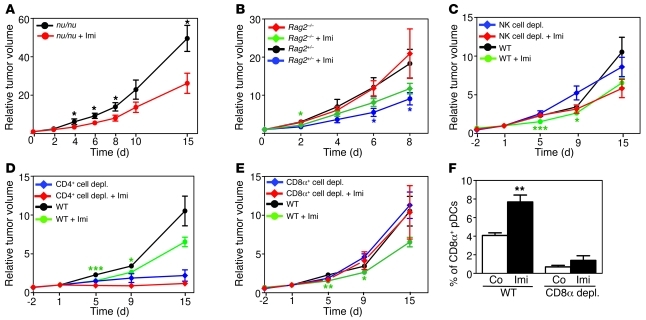
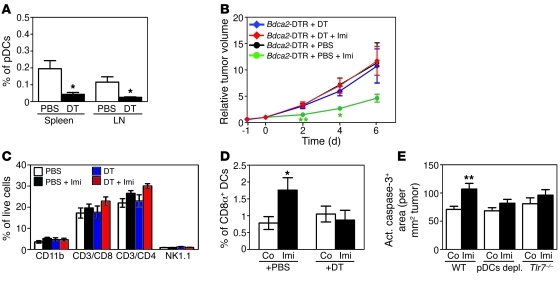
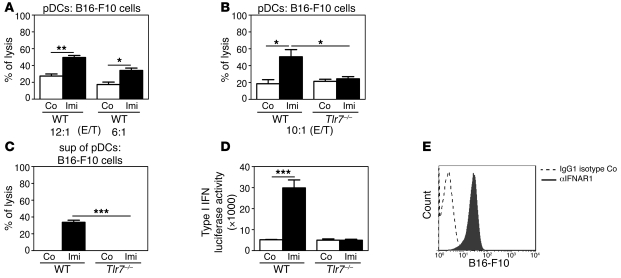
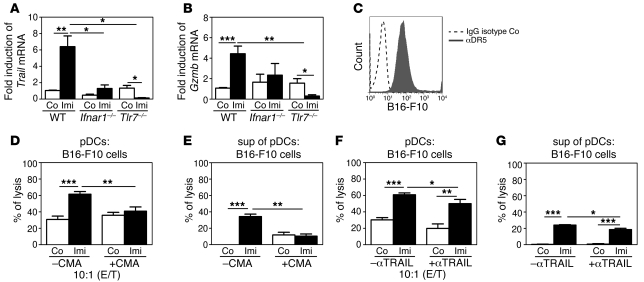
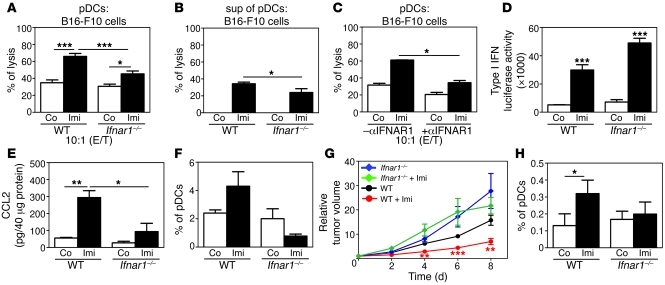
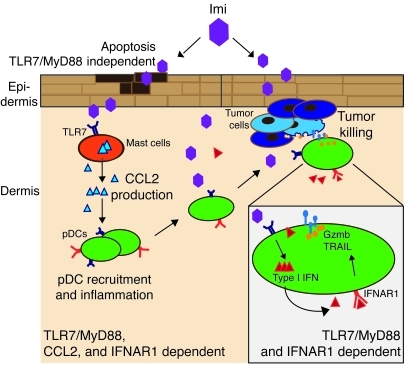
Imiquimod is a synthetic compound with antitumor properties; a 5% cream formulation is successfully used to treat skin tumors. The antitumor effect of imiquimod is multifactorial, although its ability to modulate immune responses by triggering TLR7/8 is thought to be key. Among the immune cells suggested to be involved are plasmacytoid DCs (pDCs). However, a direct contribution of pDCs to tumor killing in vivo and the mechanism of their recruitment to imiquimod-treated sites have never been demonstrated. Using a mouse model of melanoma, we have now demonstrated that pDCs can directly clear tumors without the need for the adaptive immune system. Topical imiquimod treatment led to TLR7-dependent and IFN-α/β receptor 1-dependent (IFNAR1-dependent) upregulation of expression of the chemokine CCL2 in mast cells. This was essential to induce skin inflammation and for the recruitment of pDCs to the skin. The recruited pDCs were CD8α+ and induced tumor regression in a TLR7/MyD88- and IFNAR1-dependent manner. Lack of TLR7 and IFNAR1 or depletion of pDCs or CD8α+ cells from tumor-bearing mice completely abolished the effect of imiquimod. TLR7 was essential for imiquimod-stimulated pDCs to produce IFN-α/β, which led to TRAIL and granzyme B secretion by pDCs via IFNAR1 signaling. Blocking these cytolytic molecules impaired pDC-mediated tumor killing. Our results demonstrate that imiquimod treatment leads to CCL2-dependent recruitment of pDCs and their transformation into a subset of killer DCs able to directly eliminate tumor cells.
Figures
Comment in
-
Plasmacytoid dendritic cells lead the charge against tumors.J Clin Invest. 2012 Feb;122(2):481-4. doi: 10.1172/JCI61345. Epub 2012 Jan 17. J Clin Invest. 2012. PMID: 22251700 Free PMC article.
Similar articles
-
Plasmacytoid dendritic cells lead the charge against tumors.J Clin Invest. 2012 Feb;122(2):481-4. doi: 10.1172/JCI61345. Epub 2012 Jan 17. J Clin Invest. 2012. PMID: 22251700 Free PMC article.
-
TRAIL(+) human plasmacytoid dendritic cells kill tumor cells in vitro: mechanisms of imiquimod- and IFN-α-mediated antitumor reactivity.J Immunol. 2012 Feb 15;188(4):1583-91. doi: 10.4049/jimmunol.1102437. Epub 2012 Jan 9. J Immunol. 2012. PMID: 22231699
-
Plasmacytoid dendritic cell-derived type I interferon is crucial for the adjuvant activity of Toll-like receptor 7 agonists.Blood. 2010 Mar 11;115(10):1949-57. doi: 10.1182/blood-2009-08-238543. Epub 2010 Jan 11. Blood. 2010. PMID: 20065291 Free PMC article.
-
[New perspective in immunotherapy: local imiquimod treatment].Orv Hetil. 2010 May 9;151(19):774-83. doi: 10.1556/OH.2010.28866. Orv Hetil. 2010. PMID: 20427260 Review. Hungarian.
-
Plasmacytoid dendritic cells, a role in neoplastic prevention and progression.Eur J Clin Invest. 2015 Jan;45 Suppl 1:1-8. doi: 10.1111/eci.12363. Eur J Clin Invest. 2015. PMID: 25524580 Review.
Cited by
-
Mast Cells and Skin and Breast Cancers: A Complicated and Microenvironment-Dependent Role.Cells. 2021 Apr 23;10(5):986. doi: 10.3390/cells10050986. Cells. 2021. PMID: 33922465 Free PMC article. Review.
-
Construction and validation of immune-associated lncRNA model for predicting immune status and therapeutic reactions of triple-negative breast cancer.Am J Transl Res. 2024 Sep 15;16(9):4355-4378. doi: 10.62347/VIXN9362. eCollection 2024. Am J Transl Res. 2024. PMID: 39398616 Free PMC article.
-
Intratumoral immunization: a new paradigm for cancer therapy.Clin Cancer Res. 2014 Apr 1;20(7):1747-56. doi: 10.1158/1078-0432.CCR-13-2116. Clin Cancer Res. 2014. PMID: 24691639 Free PMC article.
-
Intravenous delivery of the toll-like receptor 7 agonist SC1 confers tumor control by inducing a CD8+ T cell response.Oncoimmunology. 2019 Apr 19;8(7):1601480. doi: 10.1080/2162402X.2019.1601480. eCollection 2019. Oncoimmunology. 2019. PMID: 31143525 Free PMC article.
-
Application of toll-like receptors (TLRs) and their agonists in cancer vaccines and immunotherapy.Front Immunol. 2023 Oct 23;14:1227833. doi: 10.3389/fimmu.2023.1227833. eCollection 2023. Front Immunol. 2023. PMID: 37936697 Free PMC article. Review.
References
-
- Li VW, Li WW, Talcott KE, Zhai AW. Imiquimod as an antiangiogenic agent. J Drugs Dermatol. 2005;4(6):708–717. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
