

Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice
- PMID: 22251704
- PMCID: PMC3266772
- DOI: 10.1172/JCI43937
Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice
Abstract
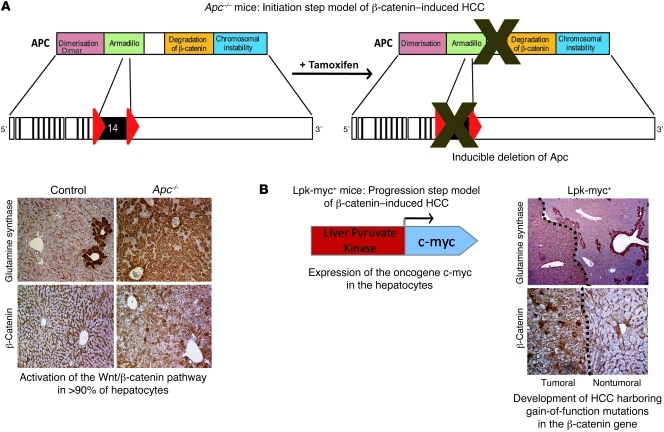
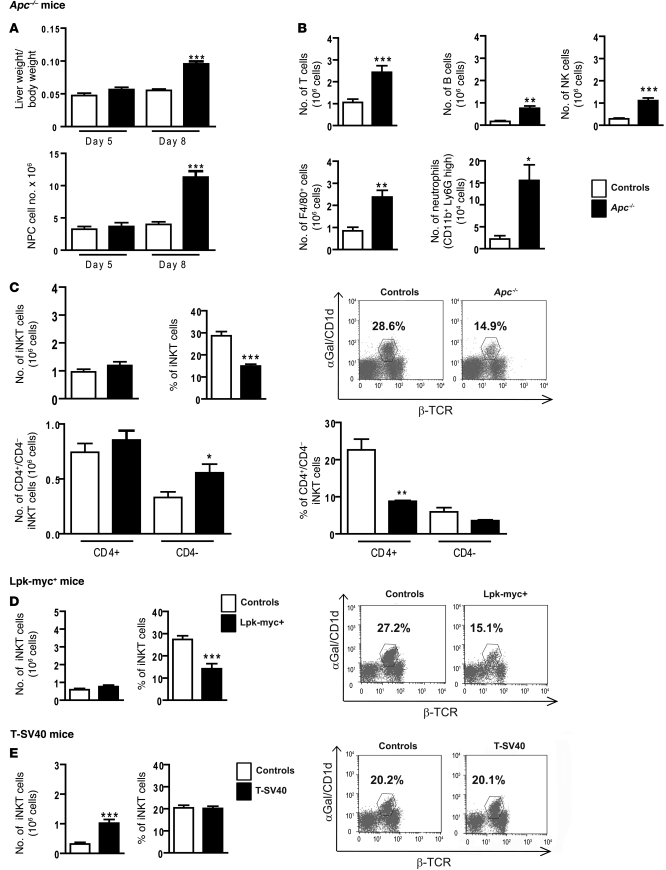
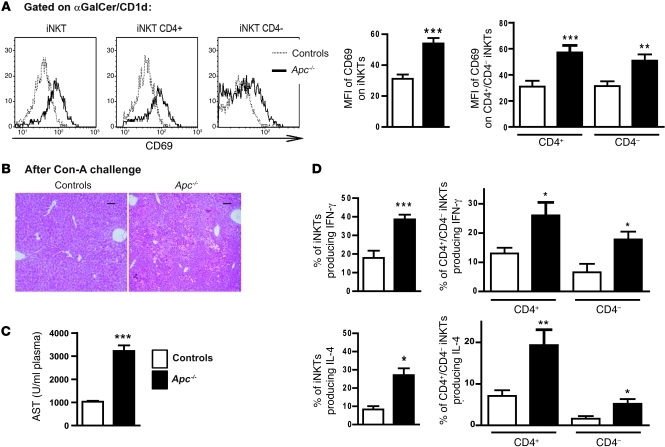
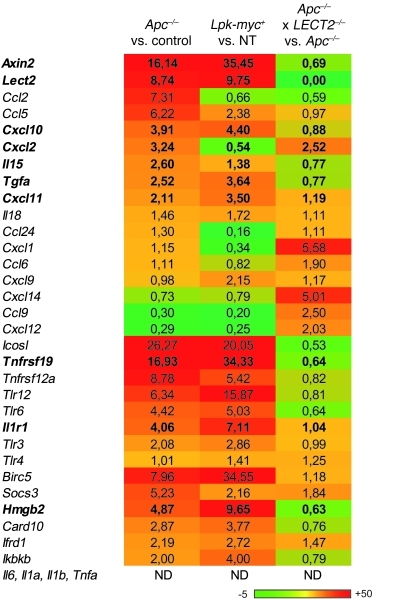
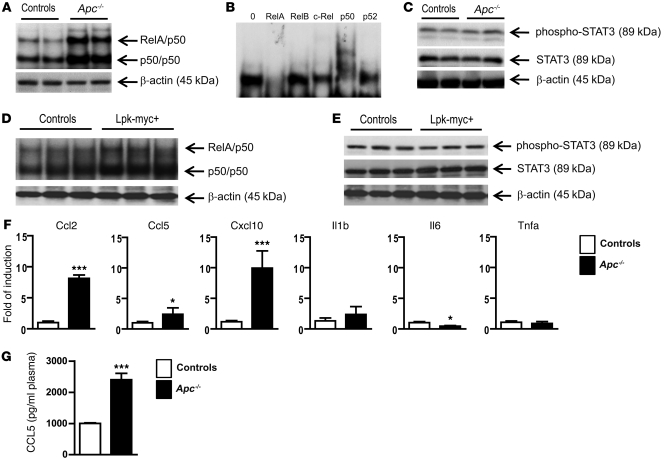
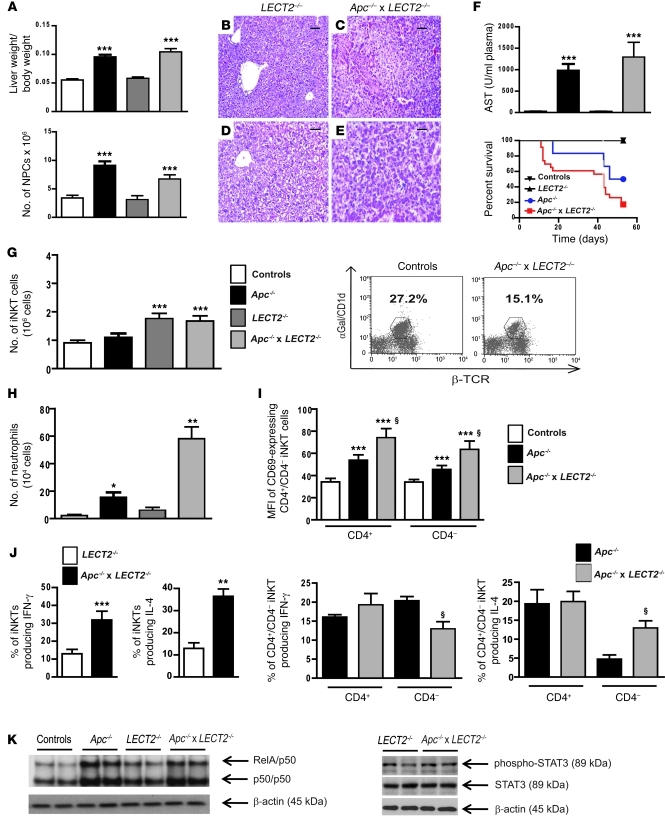
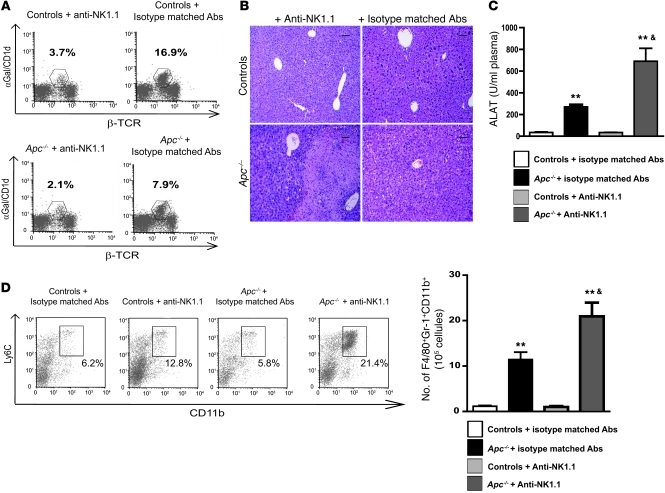
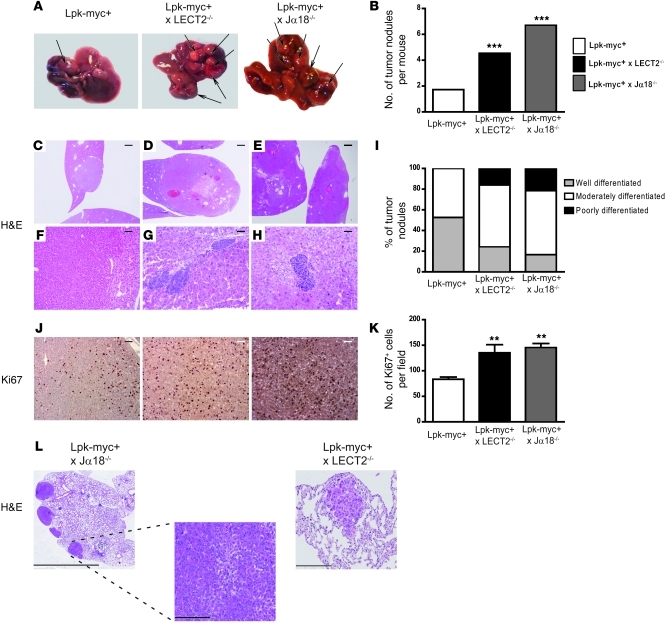
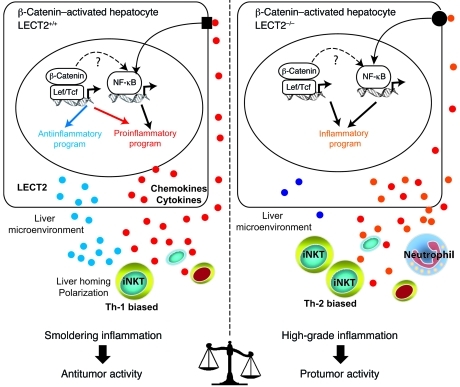
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide. Its pathogenesis is frequently linked to liver inflammation. Gain-of-function mutations in the gene encoding β-catenin are frequent genetic modifications found in human HCCs. Thus, we investigated whether inflammation was a component of β-catenin-induced tumorigenesis using genetically modified mouse models that recapitulated the stages of initiation and progression of this tumoral process. Oncogenic β-catenin signaling was found to induce an inflammatory program in hepatocytes that involved direct transcriptional control by β-catenin and activation of the NF-κB pathway. This led to a specific inflammatory response, the intensity of which determined the degree of tumor aggressiveness. The chemokine-like chemotactic factor leukocyte cell-derived chemotaxin 2 (LECT2) and invariant NKT (iNKT) cells were identified as key interconnected effectors of liver β-catenin-induced inflammation. In genetic deletion models lacking the gene encoding LECT2 or iNKT cells, hepatic β-catenin signaling triggered the formation of highly malignant HCCs with lung metastasis. Thus, our results identify inflammation as a key player in β-catenin-induced liver tumorigenesis. We provide strong evidence that, by activating pro- and antiinflammatory mediators, β-catenin signaling produces an inflammatory microenvironment that has an impact on tumoral development. Our data are consistent with the fact that most β-catenin-activated HCCs are of better prognosis.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
