

Development and mapping of SNP assays in allotetraploid cotton
- PMID: 22252442
- PMCID: PMC3324690
- DOI: 10.1007/s00122-011-1780-8
Development and mapping of SNP assays in allotetraploid cotton
Abstract
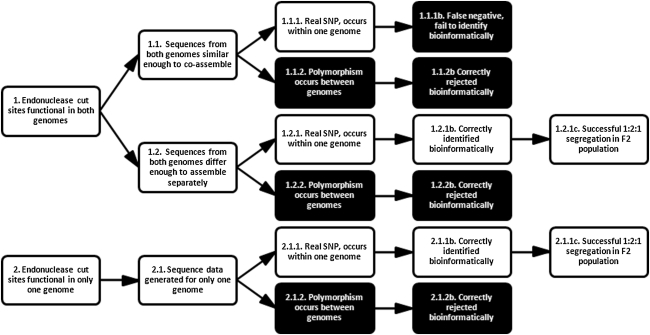
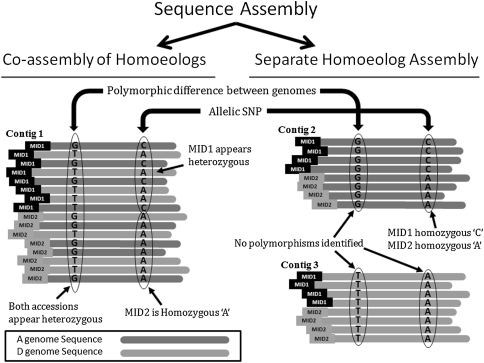
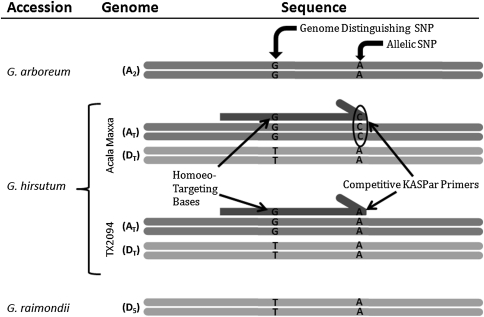
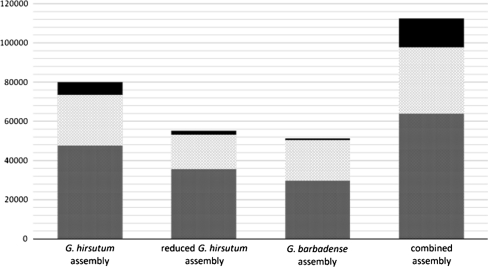
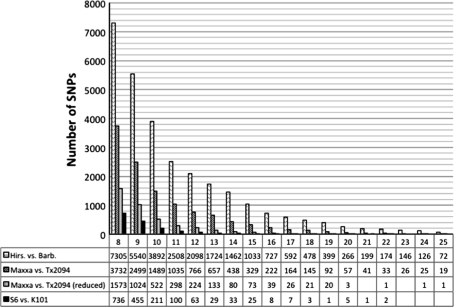
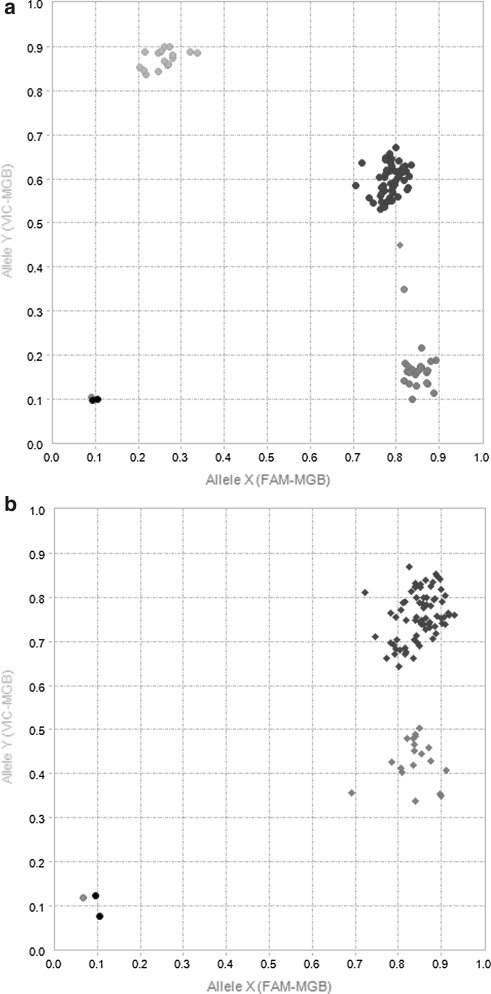
A narrow germplasm base and a complex allotetraploid genome have made the discovery of single nucleotide polymorphism (SNP) markers difficult in cotton (Gossypium hirsutum). To generate sequence for SNP discovery, we conducted a genome reduction experiment (EcoRI, BafI double digest, followed by adapter ligation, biotin-streptavidin purification, and agarose gel separation) on two accessions of G. hirsutum and two accessions of G. barbadense. From the genome reduction experiment, a total of 2.04 million genomic sequence reads were assembled into contigs with an N(50) of 508 bp and analyzed for SNPs. A previously generated assembly of expressed sequence tags (ESTs) provided an additional source for SNP discovery. Using highly conservative parameters (minimum coverage of 8× at each SNP and 20% minor allele frequency), a total of 11,834 and 1,679 non-genic SNPs were identified between accessions of G. hirsutum and G. barbadense in genome reduction assemblies, respectively. An additional 4,327 genic SNPs were also identified between accessions of G. hirsutum in the EST assembly. KBioscience KASPar assays were designed for a portion of the intra-specific G. hirsutum SNPs. From 704 non-genic and 348 genic markers developed, a total of 367 (267 non-genic, 100 genic) mapped in a segregating F(2) population (Acala Maxxa × TX2094) using the Fluidigm EP1 system. A G. hirsutum genetic linkage map of 1,688 cM was constructed based entirely on these new SNP markers. Of the genic-based SNPs, we were able to identify within which genome ('A' or 'D') each SNP resided using diploid species sequence data. Genetic maps generated by these newly identified markers are being used to locate quantitative, economically important regions within the cotton genome.
Figures

References
-
- Brubaker CL, Wendel JF. Reevaluating the origin of domesticated cotton (Gossypium hirsutum; Malvaceae) using nuclear restriction fragment length polymorphisms (RFLPs) Am J Bot. 1994;81(10):1309–1326. doi: 10.2307/2445407. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
