

Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience
- PMID: 22252923
- PMCID: PMC3278076
- DOI: 10.1002/ajmg.c.31319
Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience
Abstract
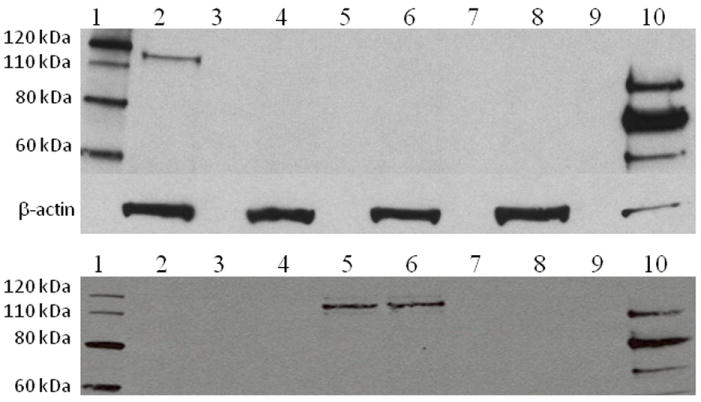
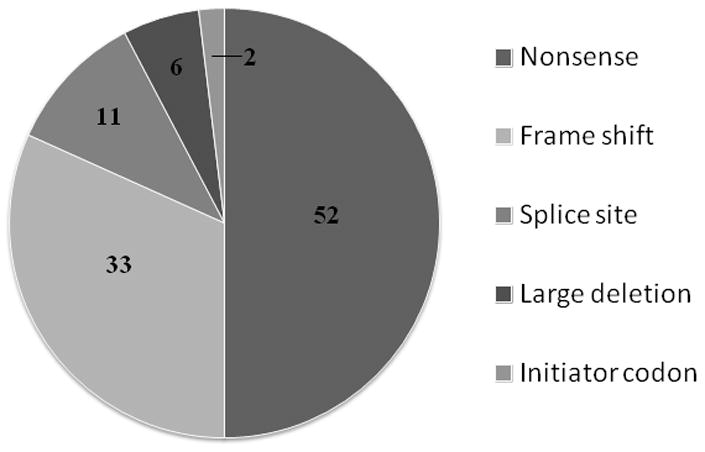
Enzyme replacement therapy (ERT) for Pompe disease using recombinant acid alpha-glucosidase (rhGAA) has resulted in increased survival although the clinical response is variable. Cross-reactive immunological material (CRIM)-negative status has been recognized as a poor prognostic factor. CRIM-negative patients make no GAA protein and develop sustained high antibody titers to ERT that render the treatment ineffective. Antibody titers are generally low for the majority of CRIM-positive patients and there is typically a better clinical outcome. Because immunomodulation has been found to be most effective in CRIM-negative patients prior to, or shortly after, initiation of ERT, knowledge of CRIM status is important before ERT is begun. We have analyzed 243 patients with infantile Pompe disease using a Western blot method for determining CRIM status and using cultured skin fibroblasts. Sixty-one out of 243 (25.1%) patients tested from various ethnic backgrounds were found to be CRIM-negative. We then correlated the CRIM results with GAA gene mutations where available (52 CRIM-negative and 88 CRIM-positive patients). We found that, in most cases, CRIM status can be predicted from GAA mutations, potentially circumventing the need for invasive skin biopsy and time wasted in culturing cells in the future. Continued studies in this area will help to increase the power of GAA gene mutations in predicting CRIM status as well as possibly identifying CRIM-positive patients who are at risk for developing high antibody titers.
Copyright © 2012 Wiley Periodicals, Inc.
Figures
References
-
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. - PubMed
-
- Amartino H, Painceira D, Pomponio RJ, Niizawa G, Sabio Paz V, Blanco M, Chamoles N. Two clinical forms of glycogen-storage disease type II in two generations of the same family. Clin Genet. 2006;69:187–188. - PubMed
-
- Angelini C, Semplicini C. Enzyme Replacement Therapy for Pompe Disease. Curr Neurol Neurosci Rep. 2011 Oct 15; Epub ahead of print. - PubMed
-
- Ausems MG, Kroos MA, Van der Kraan M, Smeitink JA, Kleijer WJ, Ploos van Amstel HK, Reuser AJ. Homozygous deletion of exon 18 leads to degradation of the lysosomal alpha-glucosidase precursor and to the infantile form of glycogen storage disease type II. Clin Genet. 1996;49:325–328. - PubMed
-
- Bali DS, Tolun AA, Goldstein JL, Dai J, Kishnani PS. Molecular analysis and protein processing in late-onset Pompe disease patients with low levels of acid alpha-glucosidase activity. Muscle Nerve. 2011;43:665–670. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
