

Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten
- PMID: 22253254
- PMCID: PMC3265635
- DOI: 10.1002/ajmg.c.31317
Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten
Abstract
Macroautophagy (often referred to as autophagy) is an evolutionarily conserved intracellular system by which macromolecules and organelles are delivered to lysosomes for degradation and recycling. Autophagy is robustly induced in response to starvation in order to generate nutrients and energy through the lysosomal degradation of cytoplasmic components. Constitutive, basal autophagy serves as a quality control mechanism for the elimination of aggregated proteins and worn-out or damaged organelles, such as mitochondria. Research during the last decade has made it clear that malfunctioning or failure of this system is associated with a wide range of human pathologies and age-related diseases. Our recent data provide strong evidence for the role of autophagy in the pathogenesis of Pompe disease, a lysosomal glycogen storage disease caused by deficiency of acid alpha-glucosidase (GAA). Large pools of autophagic debris in skeletal muscle cells can be seen in both our GAA knockout model and patients with Pompe disease. In this review, we will focus on these recent data, and comment on the not so recent observations pointing to the involvement of autophagy in skeletal muscle damage in Pompe disease.
Published 2012 Wiley Periodicals, Inc. This article is a U.S. Government work and is in the public domain in the USA.
Figures
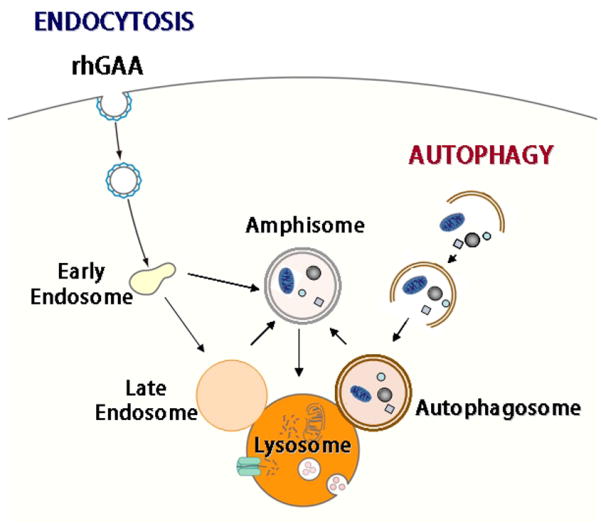

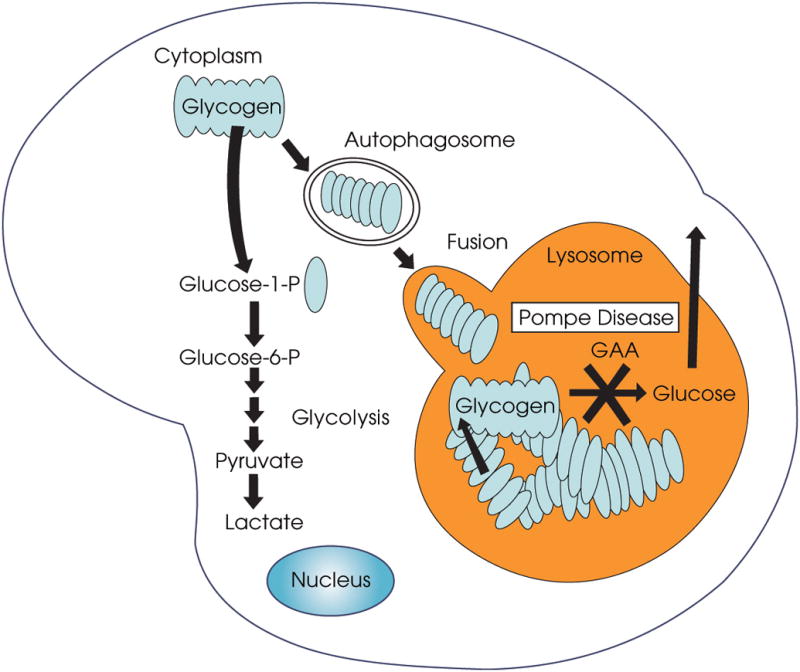


References
-
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. JBiol Chem. 1998;273:21883–21892. - PubMed
-
- Bertagnolio B, Di Donato S, Peluchetti D, Rimoldi M, Storchi G, Cornelio F. Acid maltase deficiency in adults. Clinical, morphological and biochemical study of three patients. Eur Neurol. 1978;17:193–204. - PubMed
-
- Bischoff G. Zum klinischen Bild der Glykogen-Speicherungs-Krankheit (Glykogenose) Zeitschrift fu Kinderheilkunde. 1932;52:722–725.
-
- Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Chen CA, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:e39–e45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
