

Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability
- PMID: 22253441
- PMCID: PMC3318689
- DOI: 10.1074/jbc.M111.323915
Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability
Abstract
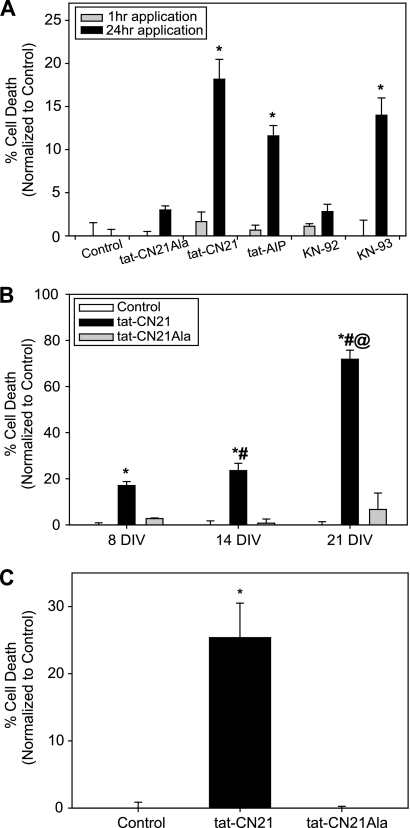
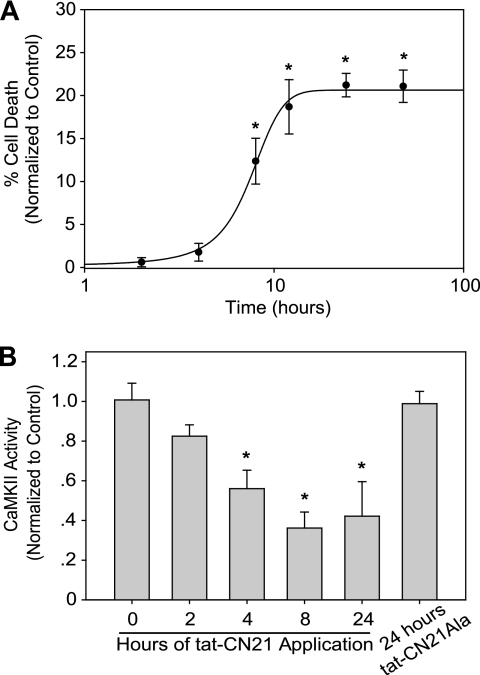
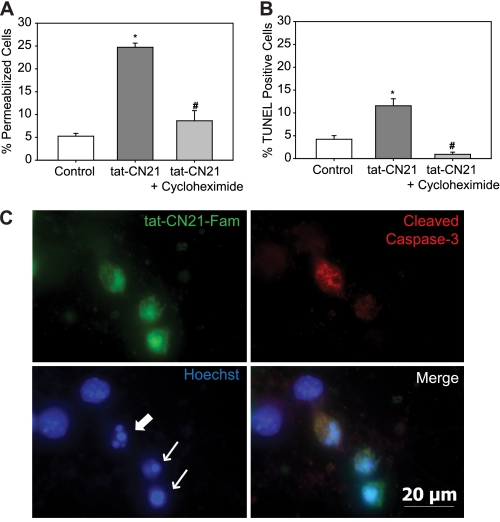
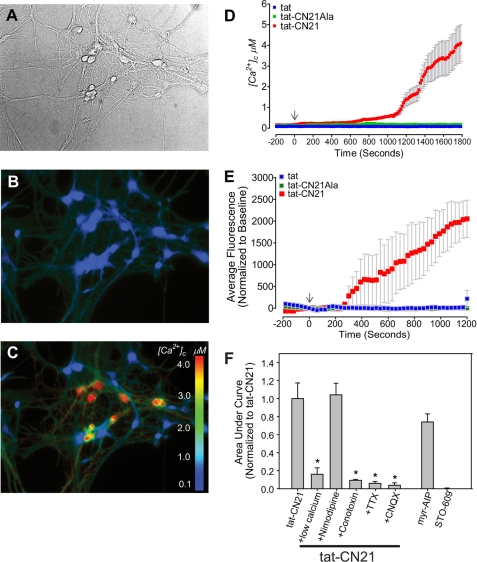
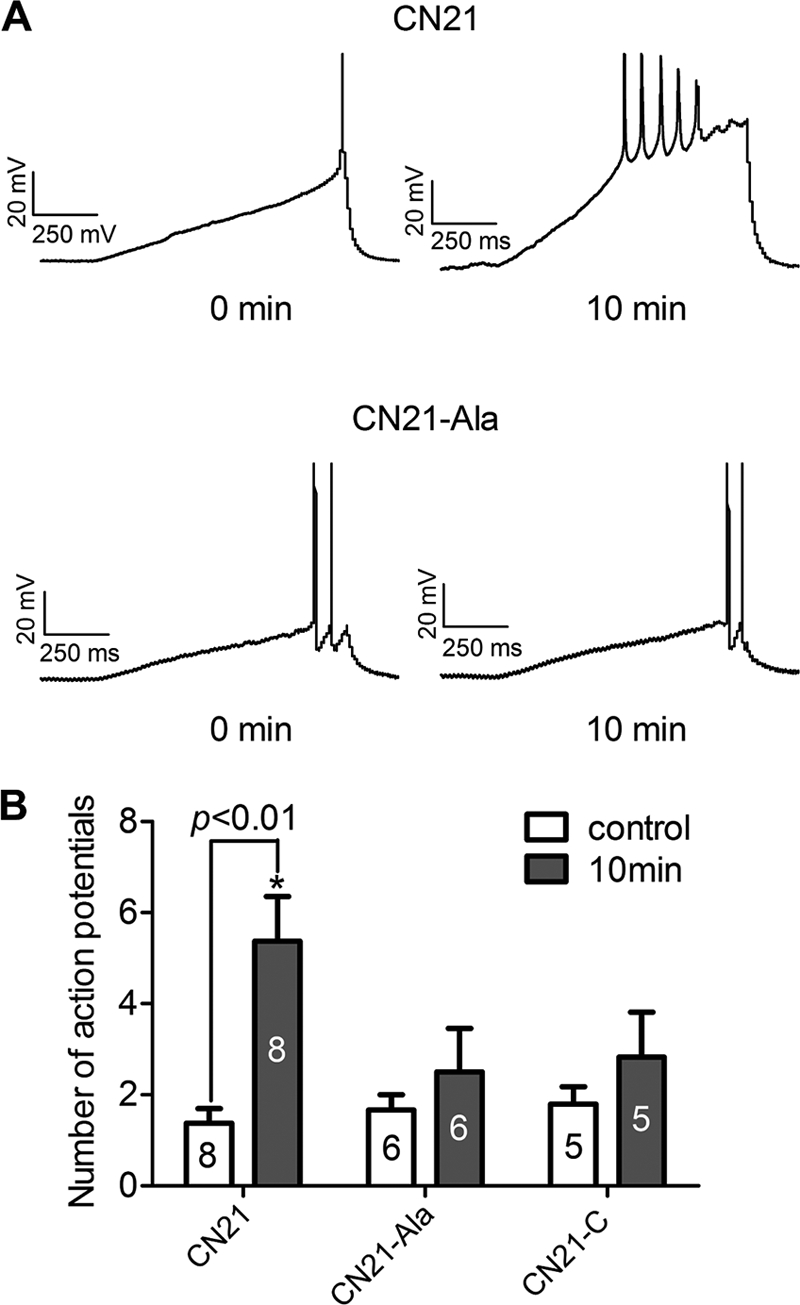
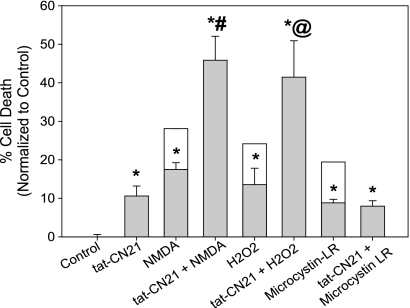
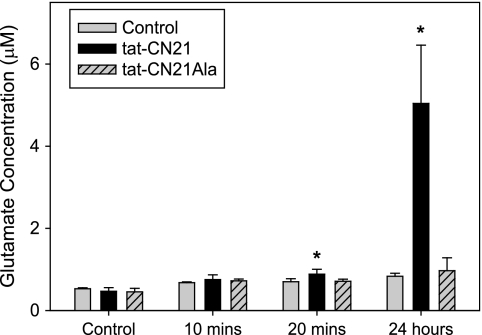
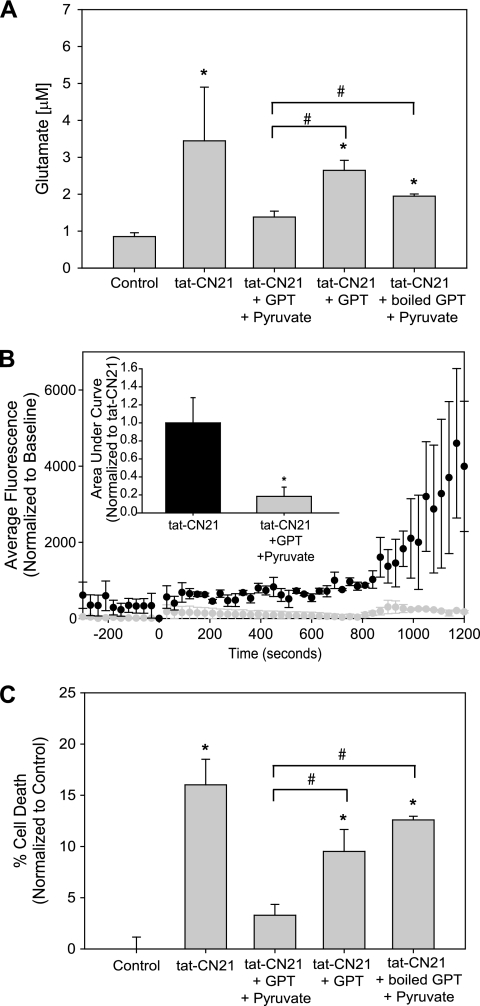
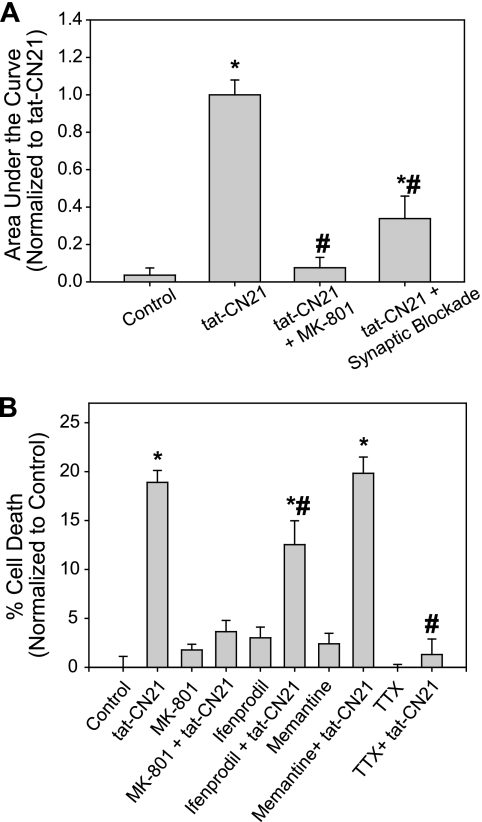
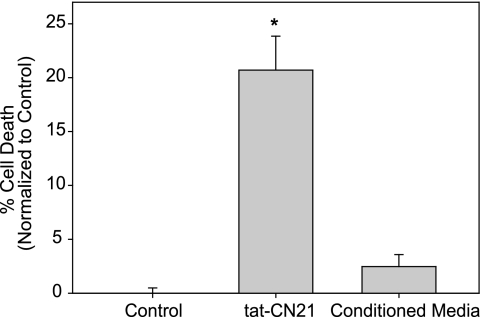
Aberrant glutamate and calcium signalings are neurotoxic to specific neuronal populations. Calcium/calmodulin-dependent kinase II (CaMKII), a multifunctional serine/threonine protein kinase in neurons, is believed to regulate neurotransmission and synaptic plasticity in response to calcium signaling produced by neuronal activity. Importantly, several CaMKII substrates control neuronal structure, excitability, and plasticity. Here, we demonstrate that CaMKII inhibition for >4 h using small molecule and peptide inhibitors induces apoptosis in cultured cortical neurons. The neuronal death produced by prolonged CaMKII inhibition is associated with an increase in TUNEL staining and caspase-3 cleavage and is blocked with the translation inhibitor cycloheximide. Thus, this neurotoxicity is consistent with apoptotic mechanisms, a conclusion that is further supported by dysregulated calcium signaling with CaMKII inhibition. CaMKII inhibitory peptides also enhance the number of action potentials generated by a ramp depolarization, suggesting increased neuronal excitability with a loss of CaMKII activity. Extracellular glutamate concentrations are augmented with prolonged inhibition of CaMKII. Enzymatic buffering of extracellular glutamate and antagonism of the NMDA subtype of glutamate receptors prevent the calcium dysregulation and neurotoxicity associated with prolonged CaMKII inhibition. However, in the absence of CaMKII inhibition, elevated glutamate levels do not induce neurotoxicity, suggesting that a combination of CaMKII inhibition and elevated extracellular glutamate levels results in neuronal death. In sum, the loss of CaMKII observed with multiple pathological states in the central nervous system, including epilepsy, brain trauma, and ischemia, likely exacerbates programmed cell death by sensitizing vulnerable neuronal populations to excitotoxic glutamate signaling and inducing an excitotoxic insult itself.
Figures
References
-
- Hudmon A., Schulman H. (2002) Neuronal Ca2+/calmodulin-dependent protein kinase II. The role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510 - PubMed
-
- Perlin J. B., Churn S. B., Lothman E. W., DeLorenzo R. J. (1992) Loss of type II calcium/calmodulin-dependent kinase activity correlates with stages of development of electrographic seizures in status epilepticus in rat. Epilepsy Res. 11, 111–118 - PubMed
-
- Zalewska T., Domanska-Janik K. (1996) Brain ischemia transiently activates Ca2+/calmodulin-independent protein kinase II. Neuroreport 7, 637–641 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
