

RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors
- PMID: 22256804
- PMCID: PMC3724537
- DOI: 10.1056/NEJMoa1105358
RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors
Abstract
Background: Cutaneous squamous-cell carcinomas and keratoacanthomas are common findings in patients treated with BRAF inhibitors.
Methods: We performed a molecular analysis to identify oncogenic mutations (HRAS, KRAS, NRAS, CDKN2A, and TP53) in the lesions from patients treated with the BRAF inhibitor vemurafenib. An analysis of an independent validation set and functional studies with BRAF inhibitors in the presence of the prevalent RAS mutation was also performed.
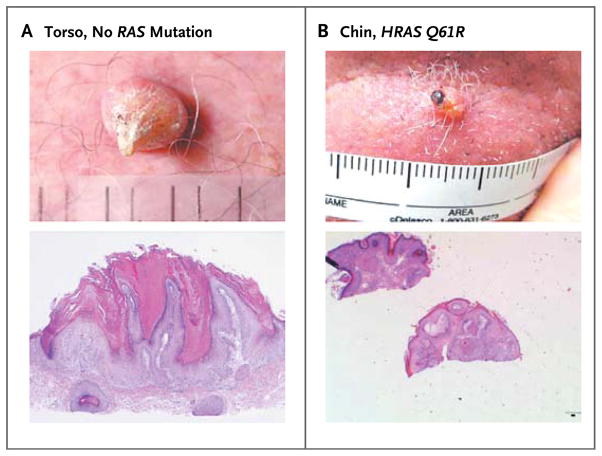
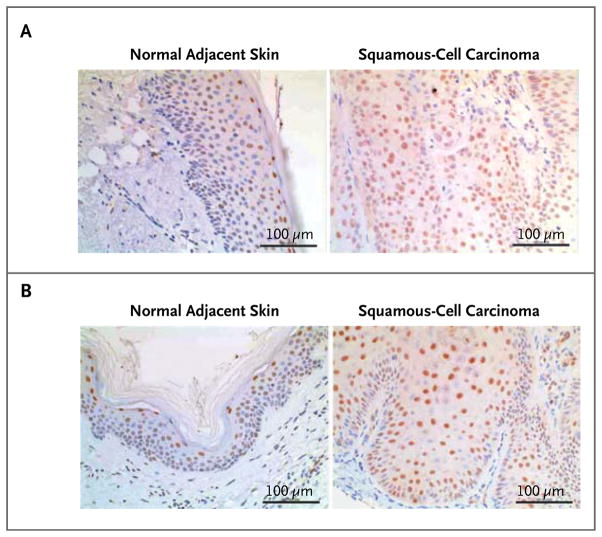
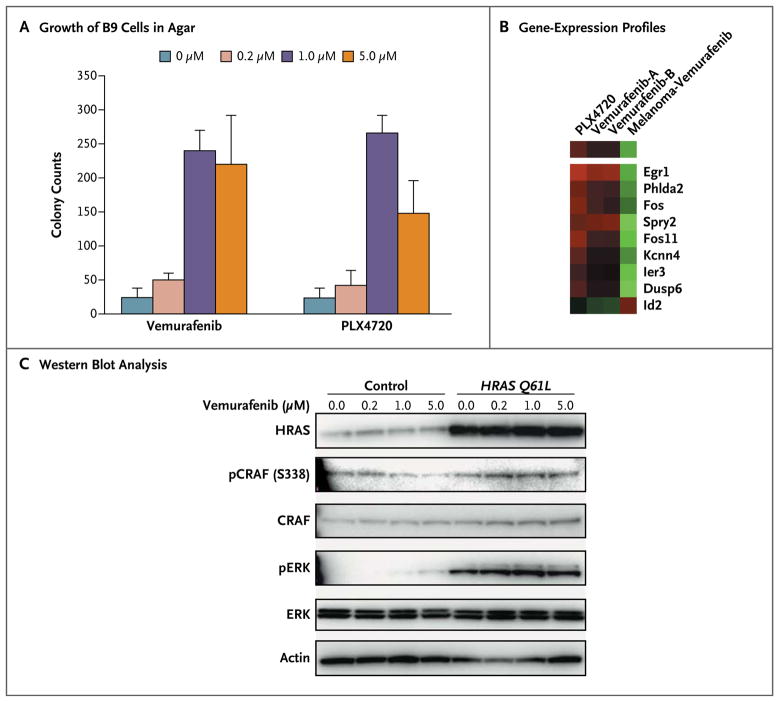
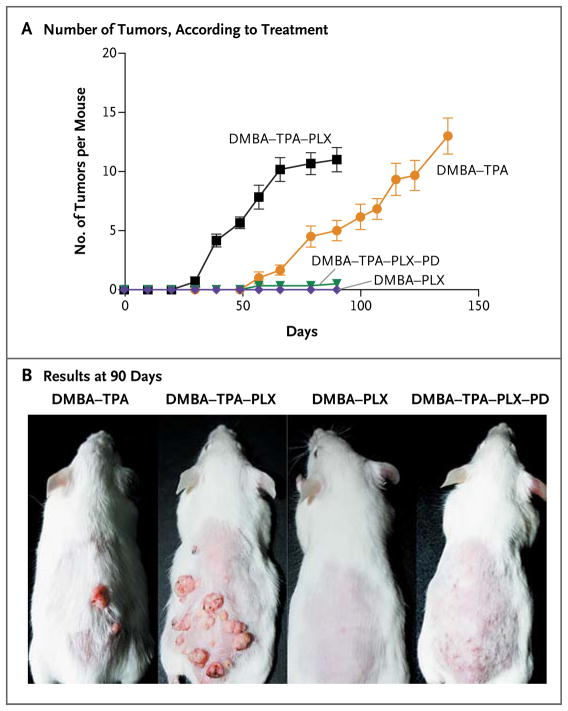
Results: Among 21 tumor samples, 13 had RAS mutations (12 in HRAS). In a validation set of 14 samples, 8 had RAS mutations (4 in HRAS). Thus, 60% (21 of 35) of the specimens harbored RAS mutations, the most prevalent being HRAS Q61L. Increased proliferation of HRAS Q61L-mutant cell lines exposed to vemurafenib was associated with mitogen-activated protein kinase (MAPK)-pathway signaling and activation of ERK-mediated transcription. In a mouse model of HRAS Q61L-mediated skin carcinogenesis, the vemurafenib analogue PLX4720 was not an initiator or a promoter of carcinogenesis but accelerated growth of the lesions harboring HRAS mutations, and this growth was blocked by concomitant treatment with a MEK inhibitor.
Conclusions: Mutations in RAS, particularly HRAS, are frequent in cutaneous squamous-cell carcinomas and keratoacanthomas that develop in patients treated with vemurafenib. The molecular mechanism is consistent with the paradoxical activation of MAPK signaling and leads to accelerated growth of these lesions. (Funded by Hoffmann-La Roche and others; ClinicalTrials.gov numbers, NCT00405587, NCT00949702, NCT01001299, and NCT01006980.).
Figures
Comment in
-
RAF around the edges--the paradox of BRAF inhibitors.N Engl J Med. 2012 Jan 19;366(3):271-3. doi: 10.1056/NEJMe1111636. N Engl J Med. 2012. PMID: 22256810 No abstract available.
References
-
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. - PubMed
-
- Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. - PubMed
-
- Ribas A, Kim K, Schuchter L, et al. BRIM-2: an open-label, multicenter Phase II study of RG7204 (PLX4032) in previously treated patients with BRAF V600E mutation-positive metastatic melanoma. J Clin Oncol. 2011;29(Suppl):8509. abstract.
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous