

The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates
- PMID: 22258098
- PMCID: PMC3379644
- DOI: 10.1038/ismej.2011.212
The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates
Abstract
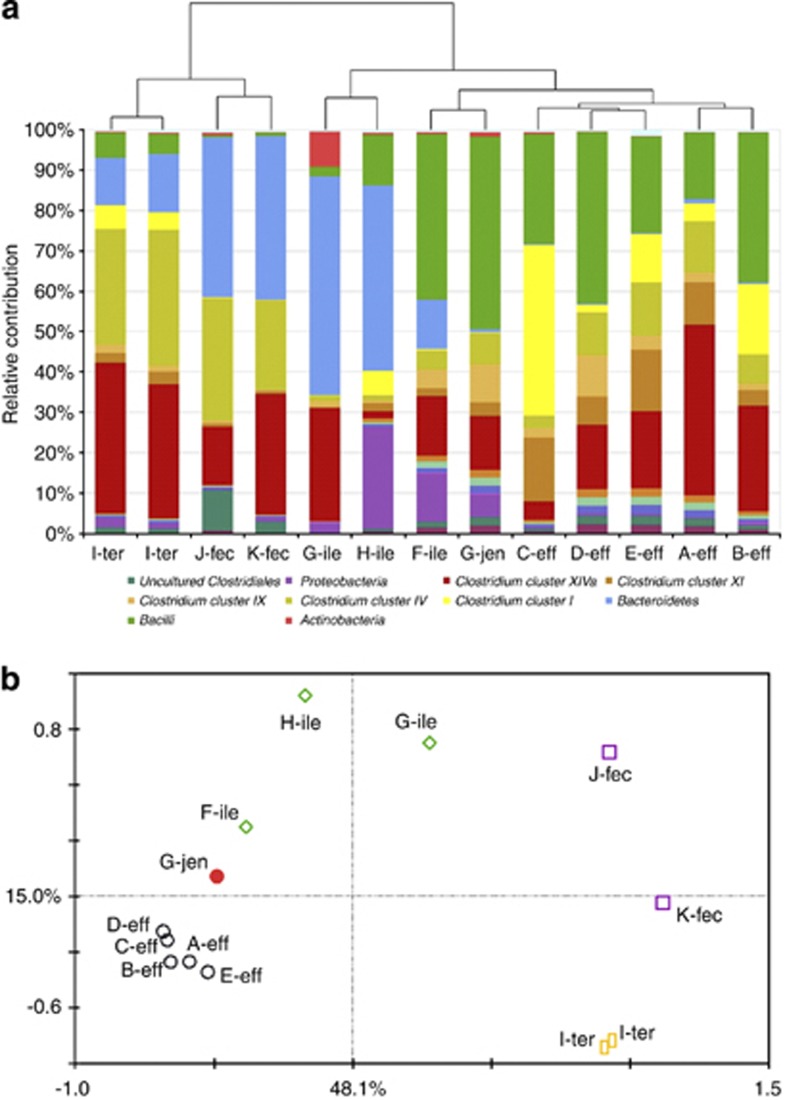
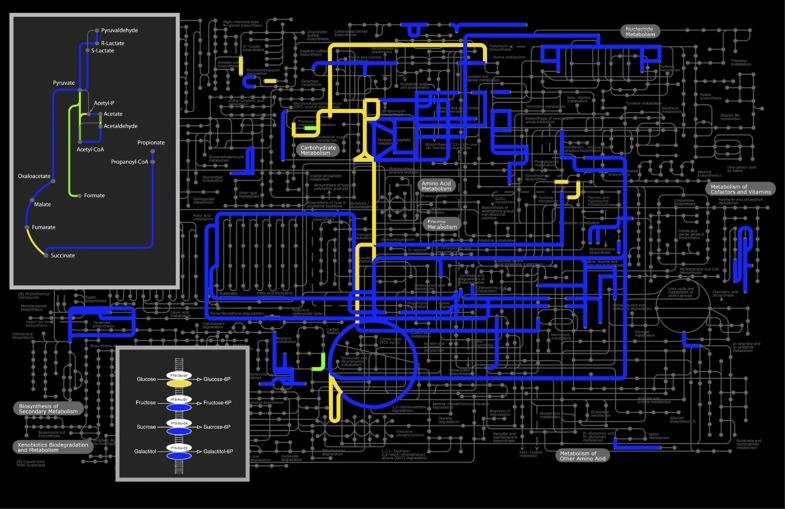
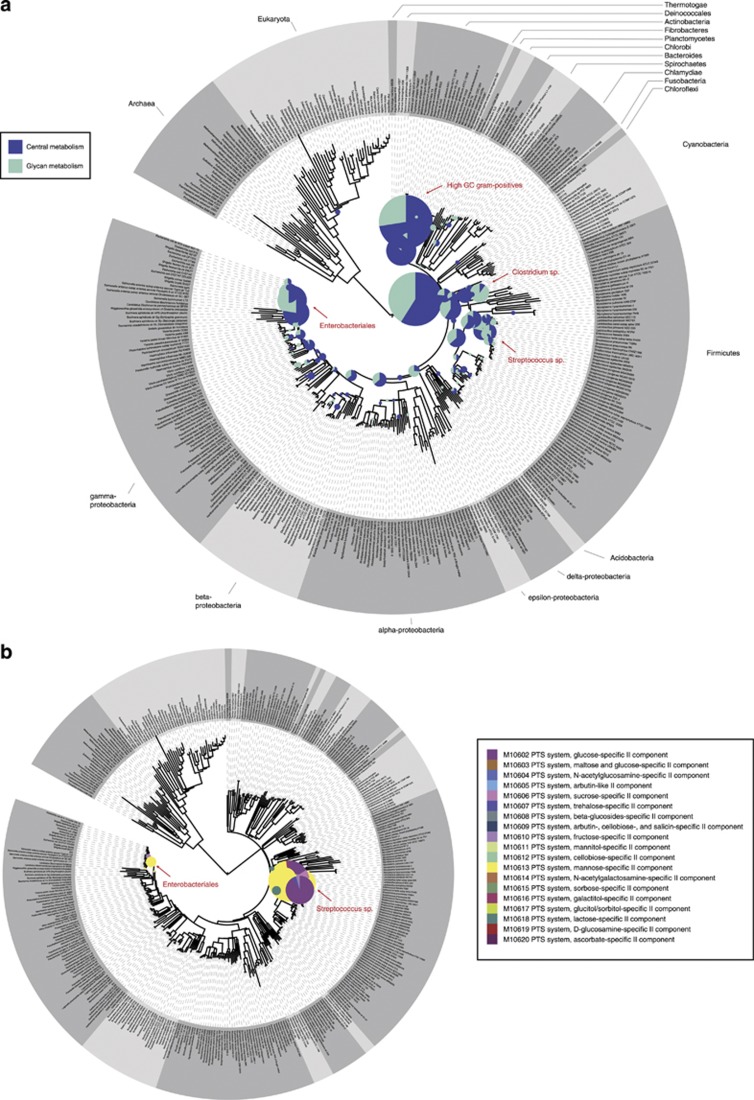
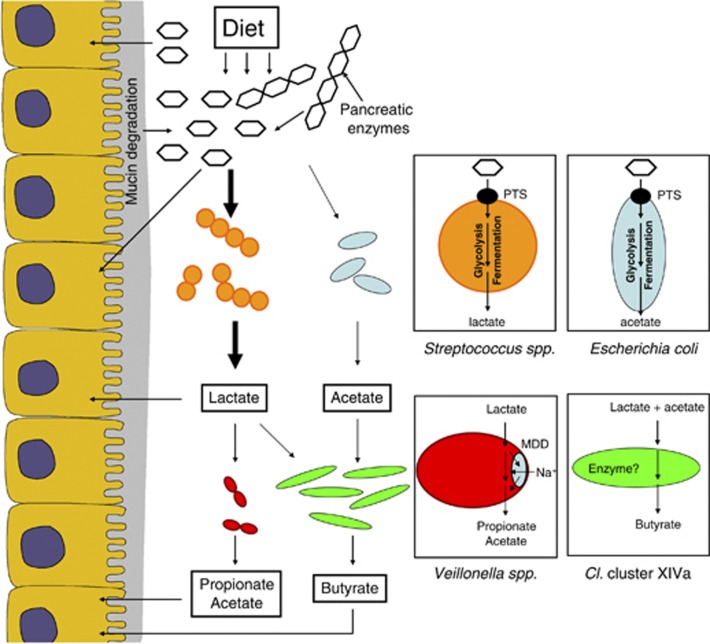
The human gastrointestinal tract (GI tract) harbors a complex community of microbes. The microbiota composition varies between different locations in the GI tract, but most studies focus on the fecal microbiota, and that inhabiting the colonic mucosa. Consequently, little is known about the microbiota at other parts of the GI tract, which is especially true for the small intestine because of its limited accessibility. Here we deduce an ecological model of the microbiota composition and function in the small intestine, using complementing culture-independent approaches. Phylogenetic microarray analyses demonstrated that microbiota compositions that are typically found in effluent samples from ileostomists (subjects without a colon) can also be encountered in the small intestine of healthy individuals. Phylogenetic mapping of small intestinal metagenome of three different ileostomy effluent samples from a single individual indicated that Streptococcus sp., Escherichia coli, Clostridium sp. and high G+C organisms are most abundant in the small intestine. The compositions of these populations fluctuated in time and correlated to the short-chain fatty acids profiles that were determined in parallel. Comparative functional analysis with fecal metagenomes identified functions that are overrepresented in the small intestine, including simple carbohydrate transport phosphotransferase systems (PTS), central metabolism and biotin production. Moreover, metatranscriptome analysis supported high level in-situ expression of PTS and carbohydrate metabolic genes, especially those belonging to Streptococcus sp. Overall, our findings suggest that rapid uptake and fermentation of available carbohydrates contribute to maintaining the microbiota in the human small intestine.
Figures
References
-
- Arumugam M, Harrington ED, Foerstner KU, Raes J, Bork P. SmashCommunity: a metagenomic annotation and analysis tool. Bioinformatics. 2010;26 (23:2977–2978. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
