

The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ
- PMID: 22258260
- PMCID: PMC3302542
- DOI: 10.1128/JVI.06778-11
The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ
Abstract
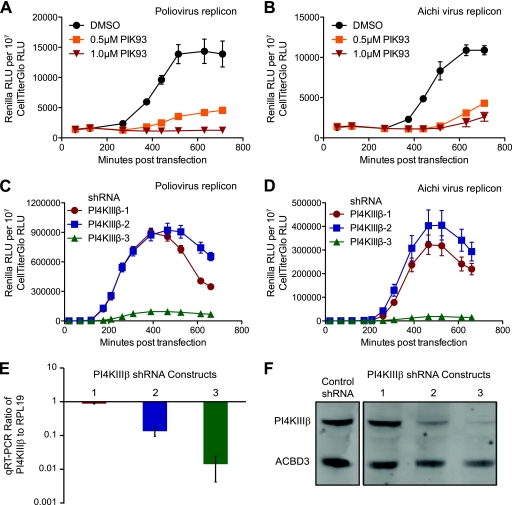
The activity of phosphatidylinositol 4-kinase class III beta (PI4KIIIβ) has been shown to be required for the replication of multiple picornaviruses; however, it is unclear whether a physical association between PI4KIIIβ and the viral replication machinery exists and, if it does, whether association is necessary. We examined the ability of the 3A protein from 18 different picornaviruses to form a complex with PI4KIIIβ by affinity purification of Strep-Tagged transiently transfected constructs followed by mass spectrometry and Western blotting for putative interacting targets. We found that the 3A proteins of Aichi virus, bovine kobuvirus, poliovirus, coxsackievirus B3, and human rhinovirus 14 all copurify with PI4KIIIβ. Furthermore, we found that multiple picornavirus 3A proteins copurify with the Golgi adaptor protein acyl coenzyme A (acyl-CoA) binding domain protein 3 (ACBD3/GPC60), including those from Aichi virus, bovine kobuvirus, human rhinovirus 14, poliovirus, and coxsackievirus B2, B3, and B5. Affinity purification of ACBD3 confirmed interaction with multiple picornaviral 3A proteins and revealed the ability to bind PI4KIIIβ in the absence of 3A. Mass-spectrometric analysis of transiently expressed Aichi virus, bovine kobuvirus, and human klassevirus 3A proteins demonstrated that the N-terminal glycines of these 3A proteins are myristoylated. Alanine-scanning mutagenesis along the entire length of Aichi virus 3A followed by transient expression and affinity purification revealed that copurification of PI4KIIIβ could be eliminated by mutation of specific residues, with little or no effect on recruitment of ACBD3. One mutation at the N terminus, I5A, significantly reduced copurification of both ACBD3 and PI4KIIIβ. The dependence of Aichi virus replication on the activity of PI4KIIIβ was confirmed by both chemical and genetic inhibition. Knockdown of ACBD3 by small interfering RNA (siRNA) also prevented replication of both Aichi virus and poliovirus. Point mutations in 3A that eliminate PI4KIIIβ association sensitized Aichi virus to PIK93, suggesting that disruption of the 3A/ACBD3/PI4KIIIβ complex may represent a novel target for therapeutic intervention that would be complementary to the inhibition of the kinase activity itself.
Figures







References
-
- Bologna G, Yvon C, Duvaud S, Veuthey A-L. 2004. N-Terminal myristoylation predictions by ensembles of neural networks. Proteomics 4:1626–1632 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
