

Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities
- PMID: 22258914
- PMCID: PMC3282414
- DOI: 10.1101/cshperspect.a009886
Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities
Abstract
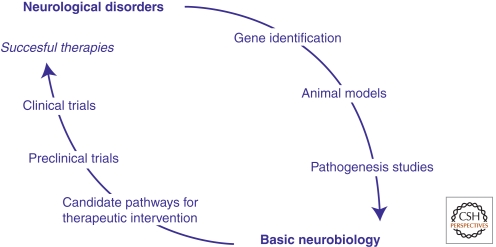
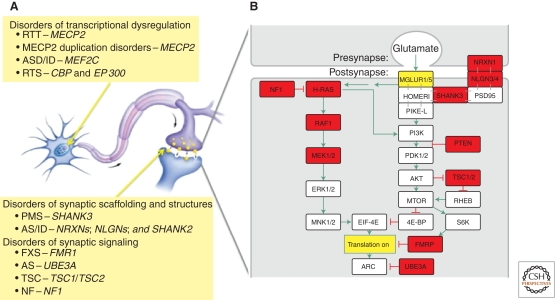
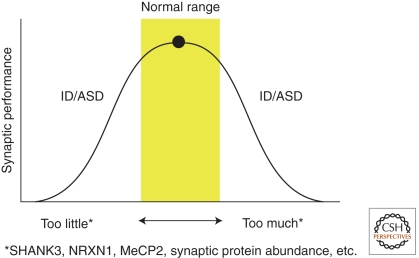
The discovery of the genetic causes of syndromic autism spectrum disorders and intellectual disabilities has greatly informed our understanding of the molecular pathways critical for normal synaptic function. The top-down approaches using human phenotypes and genetics helped identify causative genes and uncovered the broad spectrum of neuropsychiatric features that can result from various mutations in the same gene. Importantly, the human studies unveiled the exquisite sensitivity of cognitive function to precise levels of many diverse proteins. Bottom-up approaches applying molecular, biochemical, and neurophysiological studies to genetic models of these disorders revealed unsuspected pathogenic mechanisms and identified potential therapeutic targets. Moreover, studies in model organisms showed that symptoms of these devastating disorders can be reversed, which brings hope that affected individuals might benefit from interventions even after symptoms set in. Scientists predict that insights gained from studying these rare syndromic disorders will have an impact on the more common nonsyndromic autism and mild cognitive deficits.
Figures
References
-
- Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, Hudson R, Gorwill H, Nolin SL, Glicksman A, et al. 1999. Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in Fragile X study—preliminary data. Am J Med Genet 83: 322–325 - PMC - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188 - PubMed
-
- Anderlid BM, Schoumans J, Anneren G, Tapia-Paez I, Dumanski J, Blennow E, Nordenskjold M 2002. FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet 110: 439–443 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources