

High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach
- PMID: 22260654
- PMCID: PMC3368722
- DOI: 10.1186/1471-2164-13-32
High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach
Abstract
Background: Next-Generation Sequencing (NGS) is increasingly being used as a molecular epidemiologic tool for discerning ancestry and traceback of the most complicated, difficult to resolve bacterial pathogens. Making a linkage between possible food sources and clinical isolates requires distinguishing the suspected pathogen from an environmental background and placing the variation observed into the wider context of variation occurring within a serovar and among other closely related foodborne pathogens. Equally important is the need to validate these high resolution molecular tools for use in molecular epidemiologic traceback. Such efforts include the examination of strain cluster stability as well as the cumulative genetic effects of sub-culturing on these clusters. Numerous isolates of S. Montevideo were shot-gun sequenced including diverse lineage representatives as well as numerous replicate clones to determine how much variability is due to bias, sequencing error, and or the culturing of isolates. All new draft genomes were compared to 34 S. Montevideo isolates previously published during an NGS-based molecular epidemiological case study.
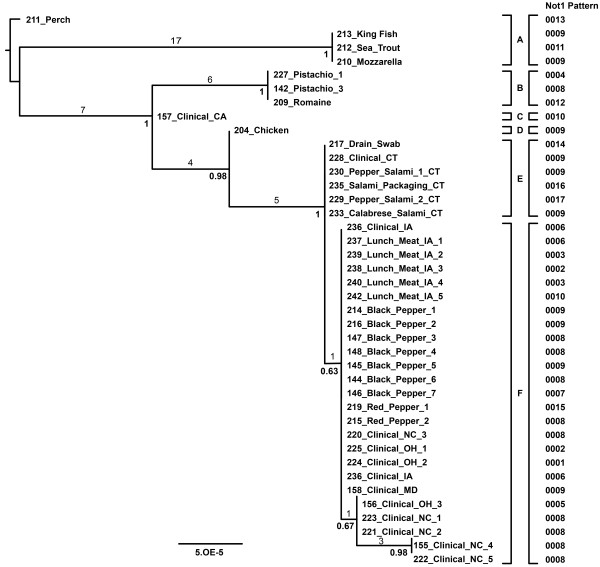
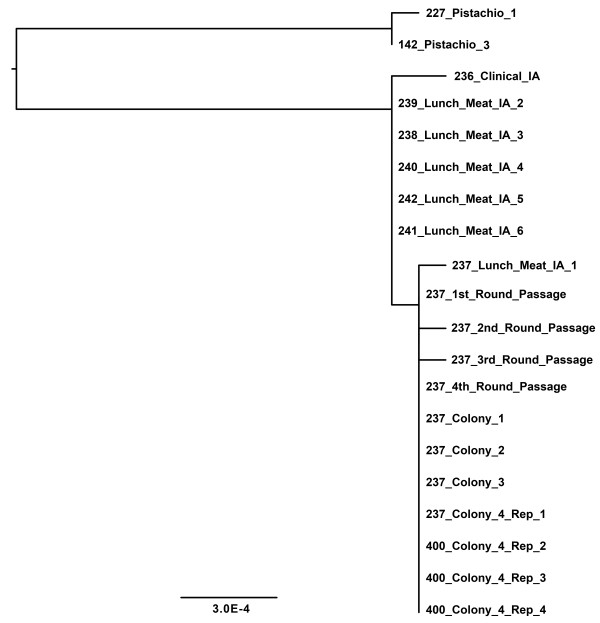
Results: Intraserovar lineages of S. Montevideo differ by thousands of SNPs, that are only slightly less than the number of SNPs observed between S. Montevideo and other distinct serovars. Much less variability was discovered within an individual S. Montevideo clade implicated in a recent foodborne outbreak as well as among individual NGS replicates. These findings were similar to previous reports documenting homopolymeric and deletion error rates with the Roche 454 GS Titanium technology. In no case, however, did variability associated with sequencing methods or sample preparations create inconsistencies with our current phylogenetic results or the subsequent molecular epidemiological evidence gleaned from these data.
Conclusions: Implementation of a validated pipeline for NGS data acquisition and analysis provides highly reproducible results that are stable and predictable for molecular epidemiological applications. When draft genomes are collected at 15×-20× coverage and passed through a quality filter as part of a data analysis pipeline, including sub-passaged replicates defined by a few SNPs, they can be accurately placed in a phylogenetic context. This reproducibility applies to all levels within and between serovars of Salmonella suggesting that investigators using these methods can have confidence in their conclusions.
Figures



References
-
- Buzby JC, Roberts T. The economics of enteric infections: human foodborne disease costs. Gastroenterology. 2009;136 1851-62.8. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
