

SSE: a nucleotide and amino acid sequence analysis platform
- PMID: 22264264
- PMCID: PMC3292810
- DOI: 10.1186/1756-0500-5-50
SSE: a nucleotide and amino acid sequence analysis platform
Abstract
Background: There is an increasing need to develop bioinformatic tools to organise and analyse the rapidly growing amount of nucleotide and amino acid sequence data in organisms ranging from viruses to eukaryotes.
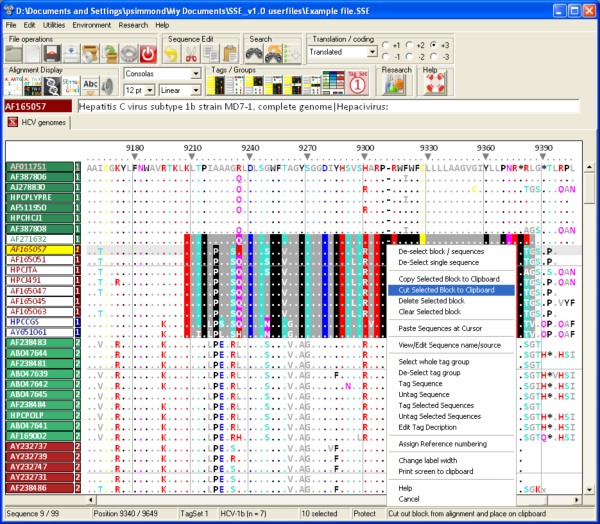
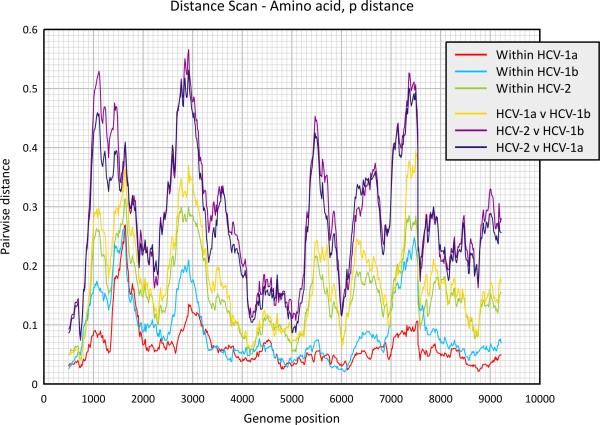
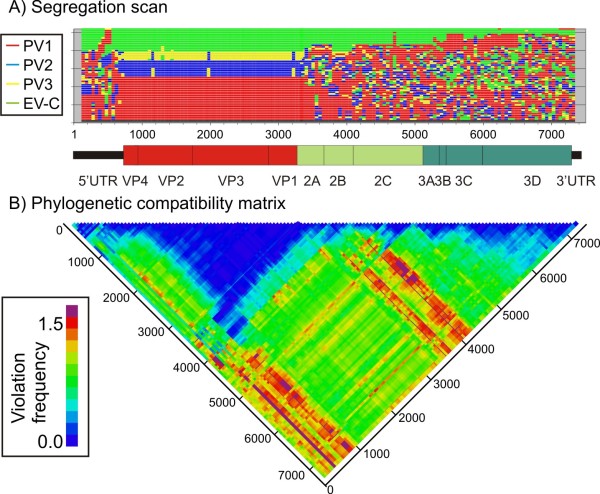
Finding: A simple sequence editor (SSE) was developed to create an integrated environment where sequences can be aligned, annotated, classified and directly analysed by a number of built-in bioinformatic programs. SSE incorporates a sequence editor for the creation of sequence alignments, a process assisted by integrated CLUSTAL/MUSCLE alignment programs and automated removal of indels. Sequences can be fully annotated and classified into groups and annotated of sequences and sequence groups and access to analytical programs that analyse diversity, recombination and RNA secondary structure. Methods for analysing sequence diversity include measures of divergence and evolutionary distances, identity plots to detect regions of nucleotide or amino acid homology, reconstruction of sequence changes, mono-, di- and higher order nucleotide compositional biases and codon usage.Association Index calculations, GroupScans, Bootscanning and TreeOrder scans perform phylogenetic analyses that reconcile group membership with tree branching orders and provide powerful methods for examining segregation of alleles and detection of recombination events. Phylogeny changes across alignments and scoring of branching order differences between trees using the Robinson-Fould algorithm allow effective visualisation of the sites of recombination events.RNA secondary and tertiary structures play important roles in gene expression and RNA virus replication. For the latter, persistence of infection is additionally associated with pervasive RNA secondary structure throughout viral genomic RNA that modulates interactions with innate cell defences. SSE provides several programs to scan alignments for RNA secondary structure through folding energy thermodynamic calculations and phylogenetic methods (detection of co-variant changes, and structure conservation between divergent sequences). These analyses complement methods based on detection of sequence constraints, such as suppression of synonymous site variability.For each program, results can be plotted in real time during analysis through an integrated graphics package, providing publication quality graphs. Results can be also directed to tabulated datafiles for import into spreadsheet or database programs for further analysis.
Conclusions: SSE combines sequence editor functions with analytical tools in a comprehensive and user-friendly package that assists considerably in bioinformatic and evolution research.
Figures

References
-
- Li WH, Graur D. Fundamentals of molecular evolution. Sinaur Associates, Inc; 1991.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
