

Single-cell analysis reveals that noncoding RNAs contribute to clonal heterogeneity by modulating transcription factor recruitment
- PMID: 22264825
- PMCID: PMC3288511
- DOI: 10.1016/j.molcel.2011.11.029
Single-cell analysis reveals that noncoding RNAs contribute to clonal heterogeneity by modulating transcription factor recruitment
Abstract
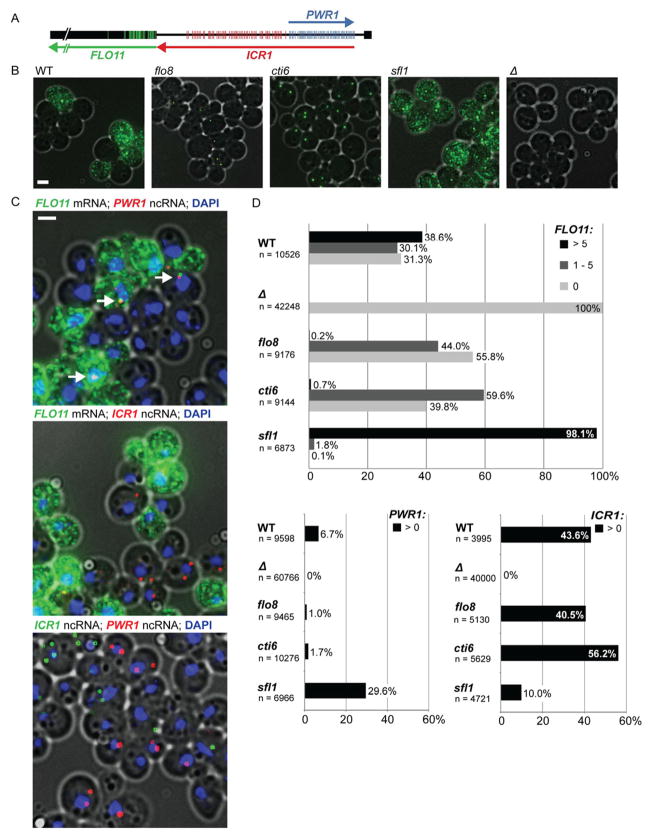
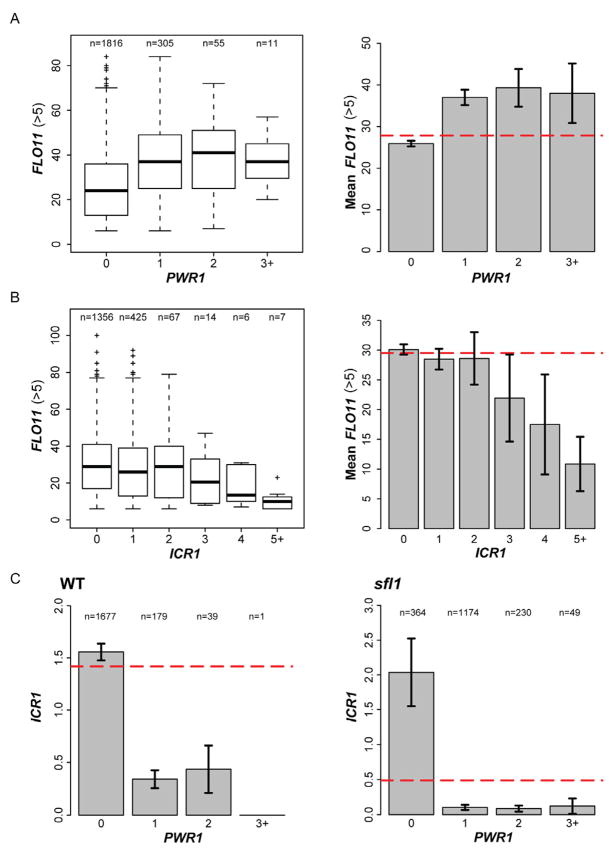
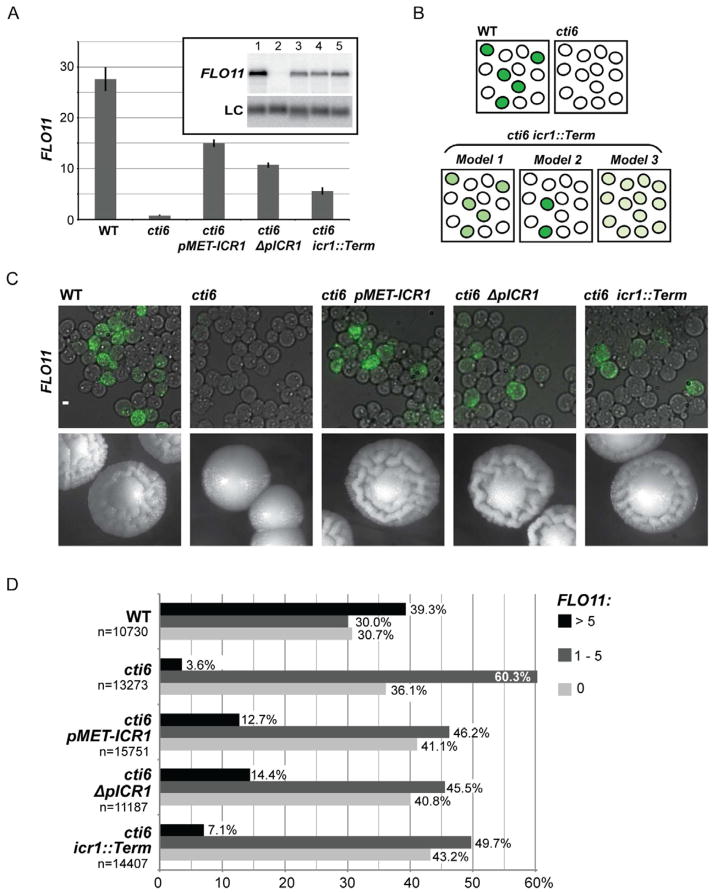
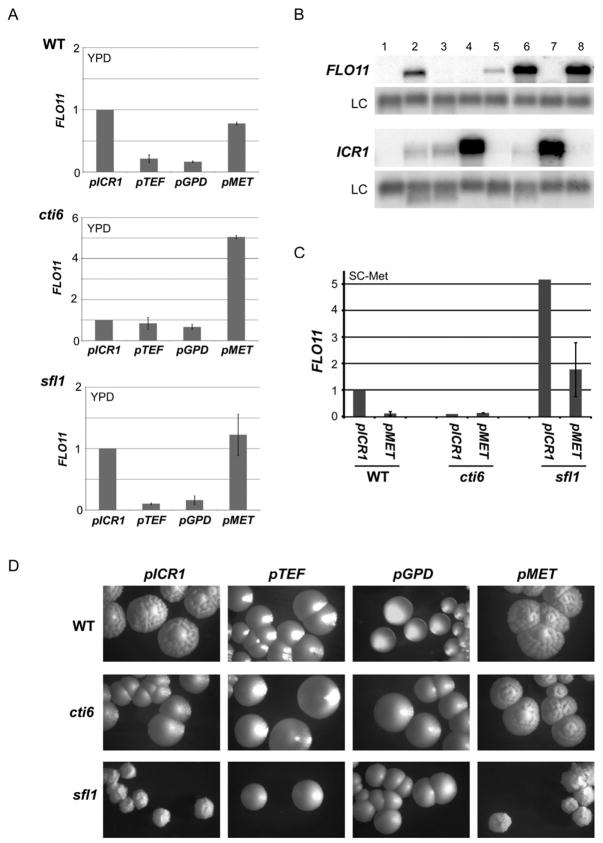
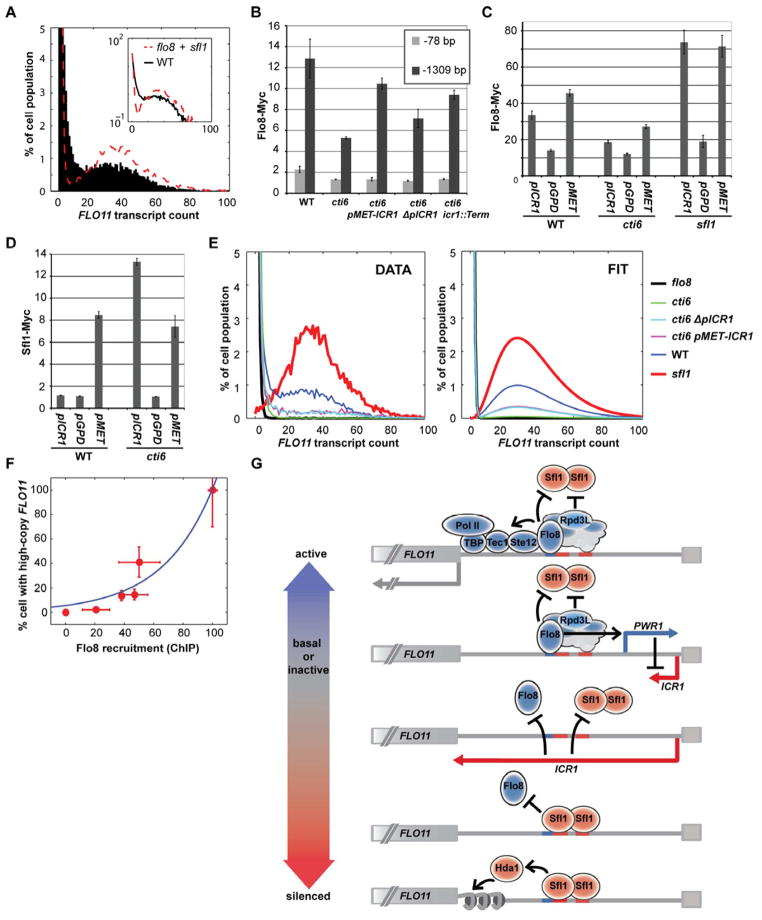
Mechanisms through which long intergenic noncoding RNAs (ncRNAs) exert regulatory effects on eukaryotic biological processes remain largely elusive. Most studies of these phenomena rely on methods that measure average behaviors in cell populations, lacking resolution to observe the effects of ncRNA transcription on gene expression in a single cell. Here, we combine quantitative single-molecule RNA FISH experiments with yeast genetics and computational modeling to gain mechanistic insights into the regulation of the Saccharomyces cerevisiae protein-coding gene FLO11 by two intergenic ncRNAs, ICR1 and PWR1. Direct detection of FLO11 mRNA and these ncRNAs in thousands of individual cells revealed alternative expression states and provides evidence that ICR1 and PWR1 contribute to FLO11's variegated transcription, resulting in Flo11-dependent phenotypic heterogeneity in clonal cell populations by modulating recruitment of key transcription factors to the FLO11 promoter.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Acar M, Mettetal JT, van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet. 2008;40:471–475. - PubMed
-
- Batada NN, Hurst LD. Evolution of chromosome organization driven by selection for reduced gene expression noise. Nat Genet. 2007;39:945–949. - PubMed
-
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
