

The p53 network: cellular and systemic DNA damage responses in aging and cancer
- PMID: 22265392
- PMCID: PMC4120491
- DOI: 10.1016/j.tig.2011.12.002
The p53 network: cellular and systemic DNA damage responses in aging and cancer
Abstract
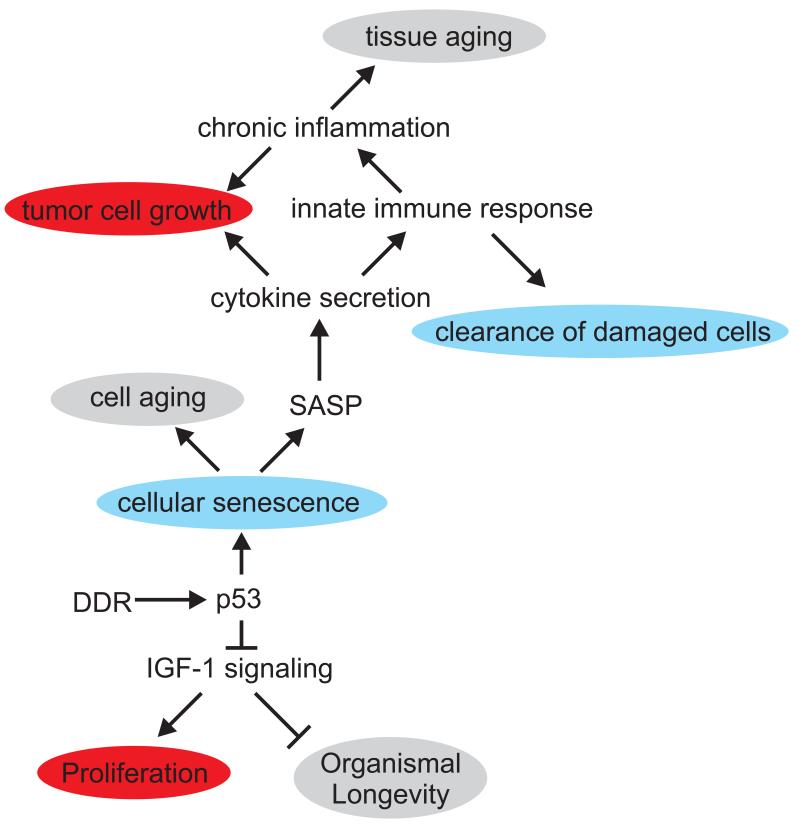
Genome instability contributes to cancer development and accelerates age-related pathologies as evidenced by a variety of congenital cancer susceptibility and progeroid syndromes that are caused by defects in genome maintenance mechanisms. DNA damage response (DDR) pathways that are mediated through the tumor suppressor p53 play an important role in the cell-intrinsic responses to genome instability, including a transient cell cycle arrest, senescence and apoptosis. Both senescence and apoptosis are powerful tumor-suppressive pathways preventing the uncontrolled proliferation of transformed cells. However, both pathways can potentially deplete stem and progenitor cell pools, thus promoting tissue degeneration and organ failure, which are both hallmarks of aging. p53 signaling is also involved in mediating non-cell-autonomous interactions with the innate immune system and in the systemic adjustments during the aging process. The network of p53 target genes thus functions as an important regulator of cancer prevention and aging.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures


References
-
- Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. - PubMed
-
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. - PubMed
-
- Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. - PubMed
-
- Bartkova J, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. - PubMed
-
- Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
