

Detecting rare variant associations by identity-by-descent mapping in case-control studies
- PMID: 22267498
- PMCID: PMC3316661
- DOI: 10.1534/genetics.111.136937
Detecting rare variant associations by identity-by-descent mapping in case-control studies
Abstract
Identity-by-descent (IBD) mapping tests whether cases share more segments of IBD around a putative causal variant than do controls. These segments of IBD can be accurately detected from genome-wide SNP data. We investigate the power of IBD mapping relative to that of SNP association testing for genome-wide case-control SNP data. Our focus is particularly on rare variants, as these tend to be more recent and hence more likely to have recent shared ancestry. We simulate data from both large and small populations and find that the relative performance of IBD mapping and SNP association testing depends on population demographic history and the strength of selection against causal variants. We also present an IBD mapping analysis of a type 1 diabetes data set. In those data we find that we can detect association only with the HLA region using IBD mapping. Overall, our results suggest that IBD mapping may have higher power than association analysis of SNP data when multiple rare causal variants are clustered within a gene. However, for outbred populations, very large sample sizes may be required for genome-wide significance unless the causal variants have strong effects.
Figures
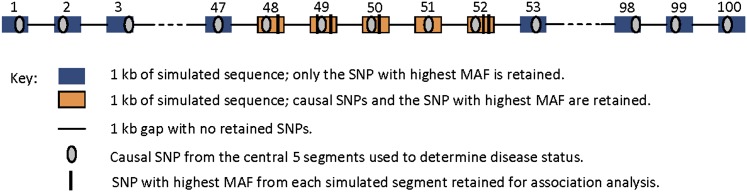
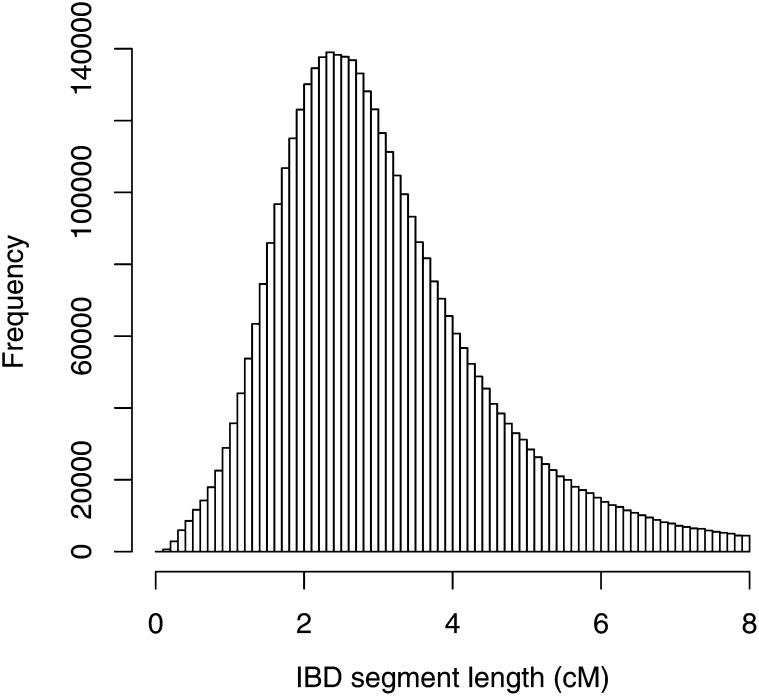
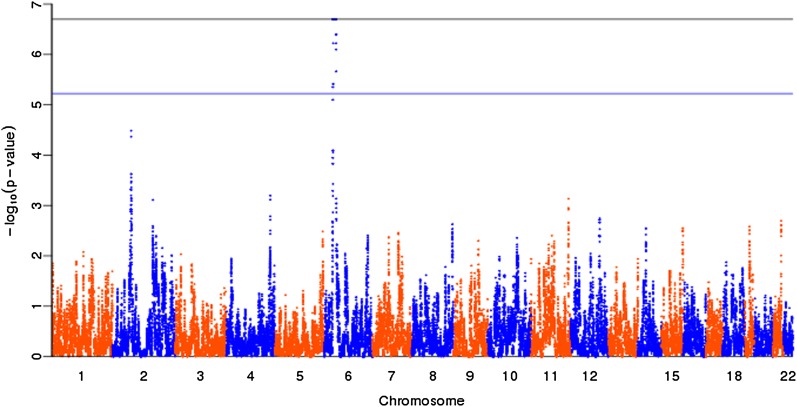
Similar articles
-
Fast pairwise IBD association testing in genome-wide association studies.Bioinformatics. 2014 Jan 15;30(2):206-13. doi: 10.1093/bioinformatics/btt609. Epub 2013 Oct 24. Bioinformatics. 2014. PMID: 24158599 Free PMC article.
-
Fast and Accurate Shared Segment Detection and Relatedness Estimation in Un-phased Genetic Data via TRUFFLE.Am J Hum Genet. 2019 Jul 3;105(1):78-88. doi: 10.1016/j.ajhg.2019.05.007. Epub 2019 Jun 6. Am J Hum Genet. 2019. PMID: 31178127 Free PMC article.
-
Detection of identity by descent using next-generation whole genome sequencing data.BMC Bioinformatics. 2012 Jun 6;13:121. doi: 10.1186/1471-2105-13-121. BMC Bioinformatics. 2012. PMID: 22672699 Free PMC article.
-
Identity by descent between distant relatives: detection and applications.Annu Rev Genet. 2012;46:617-33. doi: 10.1146/annurev-genet-110711-155534. Epub 2012 Sep 17. Annu Rev Genet. 2012. PMID: 22994355 Review.
-
Tag SNP selection for association studies.Genet Epidemiol. 2004 Dec;27(4):365-74. doi: 10.1002/gepi.20028. Genet Epidemiol. 2004. PMID: 15372618 Review.
Cited by
-
Selecting Clustering Algorithms for Identity-By-Descent Mapping.Pac Symp Biocomput. 2023;28:121-132. Pac Symp Biocomput. 2023. PMID: 36540970 Free PMC article.
-
A renewal theory approach to IBD sharing.Theor Popul Biol. 2014 Nov;97:35-48. doi: 10.1016/j.tpb.2014.08.002. Epub 2014 Aug 18. Theor Popul Biol. 2014. PMID: 25149691 Free PMC article.
-
Current Developments in Detection of Identity-by-Descent Methods and Applications.Front Genet. 2021 Sep 10;12:722602. doi: 10.3389/fgene.2021.722602. eCollection 2021. Front Genet. 2021. PMID: 34567074 Free PMC article. Review.
-
Identity-by-descent segments in large samples.bioRxiv [Preprint]. 2025 Jan 7:2024.06.05.597656. doi: 10.1101/2024.06.05.597656. bioRxiv. 2025. Update in: Theor Popul Biol. 2025 Jul 5;165:10-21. doi: 10.1016/j.tpb.2025.06.003. PMID: 38895476 Free PMC article. Updated. Preprint.
-
Mathematical bounds on r2 and the effect size in case-control genome-wide association studies.Theor Popul Biol. 2025 Aug;164:1-11. doi: 10.1016/j.tpb.2025.04.003. Epub 2025 May 15. Theor Popul Biol. 2025. PMID: 40381956
References
-
- Albrechtsen A., Korneliussen T. S., Moltle I., Hansen T. V., Nielsen F. C., et al. , 2009. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet. Epidemiol. 33(3): 266–274 - PubMed
-
- Aldous D. J., 2010. Probability Approximations via the Poisson Clumping Heuristic. Springer-Verlag, New York
-
- Baker P. R., Steck A. K., 2011. The past, present, and future of genetic associations in type 1 diabetes. Curr. Diab. Rep. 11(5): 445–453 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
