

Inhibition of AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1
- PMID: 22267730
- PMCID: PMC3318690
- DOI: 10.1074/jbc.M111.315812
Inhibition of AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1
Abstract
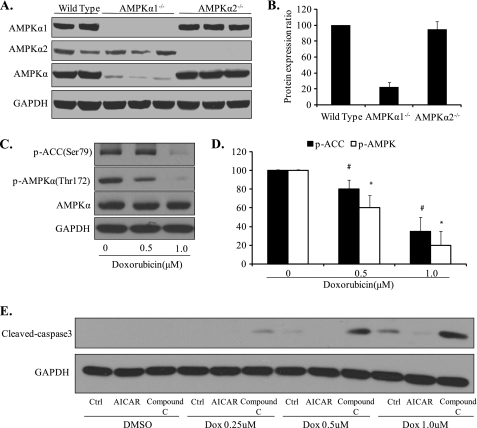
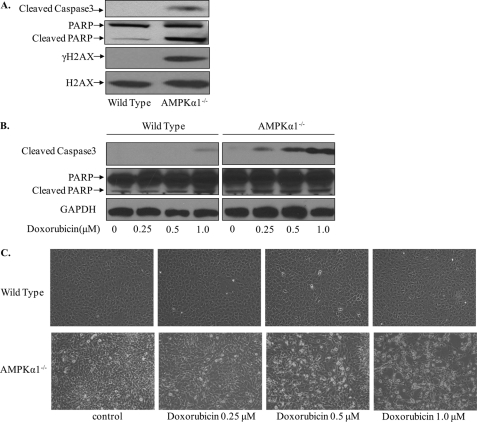
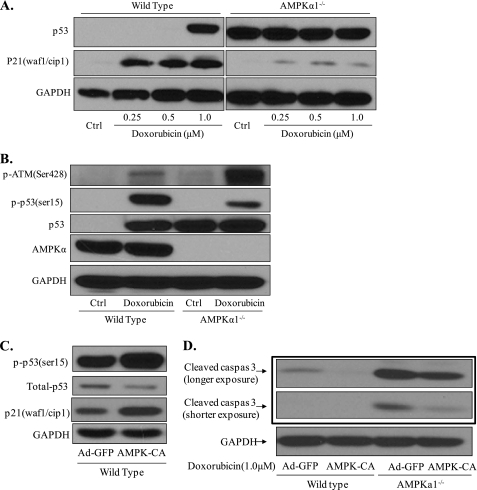
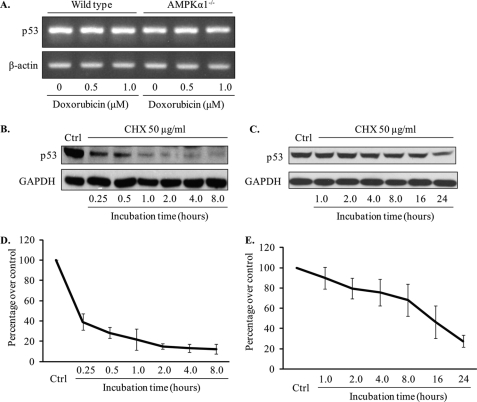
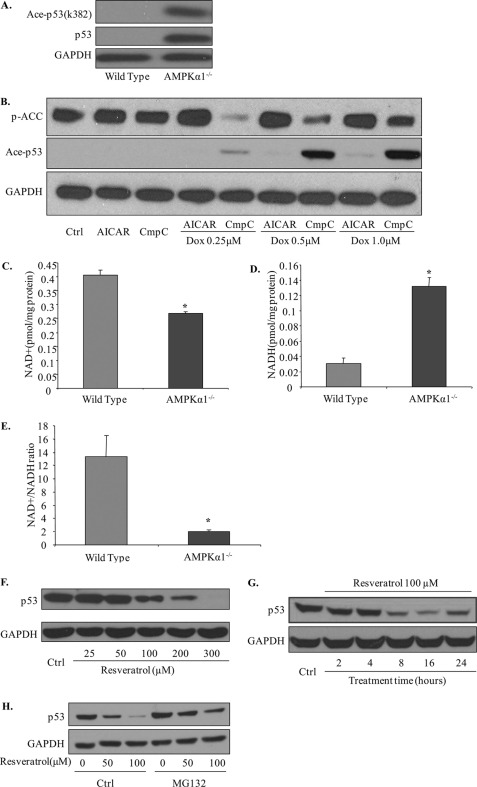
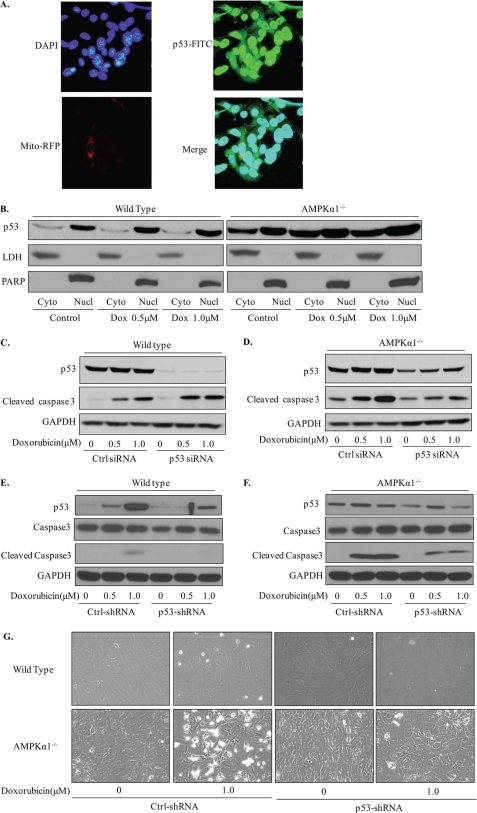
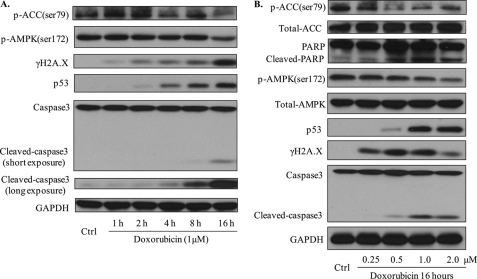
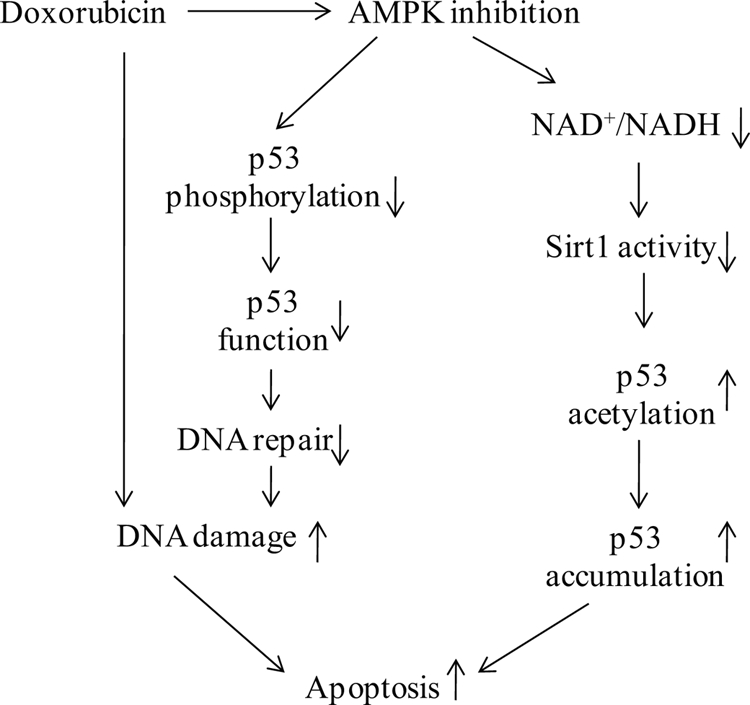
Doxorubicin, an anthracycline antibiotic, is widely used in cancer treatment. Doxorubicin produces genotoxic stress and p53 activation in both carcinoma and non-carcinoma cells. Although its side effects in non-carcinoma cells, especially in heart tissue, are well known, the molecular targets of doxorubicin are poorly characterized. Here, we report that doxorubicin inhibits AMP-activated protein kinase (AMPK) resulting in SIRT1 dysfunction and p53 accumulation. Spontaneously immortalized mouse embryonic fibroblasts (MEFs) or H9C2 cardiomyocyte were exposed to doxorubicin at different doses and durations. Cell death and p53, SIRT1, and AMPK levels were examined by Western blot. In MEFs, doxorubicin inhibited AMPK activation, increased cell death, and induced robust p53 accumulation. Genetic deletion of AMPKα1 reduced NAD(+) levels and SIRT1 activity and significantly increased the levels of p53 and cell death. Pre-activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleoside or transfection with an adenovirus encoding a constitutively active AMPK (AMPK-CA) markedly reduced the effects of doxorubicin in MEFs from Ampkα1 knock-out mice. Conversely, pre-inhibition of Ampk further sensitized MEFs to doxorubicin-induced cell death. Genetic knockdown of p53 protected both wild-type and Ampkα1(-/-) MEFs from doxorubicin-induced cell death. p53 accumulation in Ampkα1(-/-) MEFs was reversed by SIRT1 activation by resveratrol. Taken together, these data suggest that AMPK inhibition by doxorubicin causes p53 accumulation and SIRT1 dysfunction in MEFs and further suggest that pharmacological activation of AMPK might alleviate the side effects of doxorubicin.
Figures
Similar articles
-
AMP-activated protein kinase α2 and E2F1 transcription factor mediate doxorubicin-induced cytotoxicity by forming a positive signal loop in mouse embryonic fibroblasts and non-carcinoma cells.J Biol Chem. 2014 Feb 21;289(8):4839-52. doi: 10.1074/jbc.M113.496315. Epub 2014 Jan 7. J Biol Chem. 2014. PMID: 24398673 Free PMC article.
-
AICAR and compound C negatively modulate HCC-induced primary human hepatic stellate cell activation in vitro.Am J Physiol Gastrointest Liver Physiol. 2021 Apr 1;320(4):G543-G556. doi: 10.1152/ajpgi.00262.2020. Epub 2021 Jan 6. Am J Physiol Gastrointest Liver Physiol. 2021. PMID: 33406006
-
AMP-activated protein kinase suppresses matrix metalloproteinase-9 expression in mouse embryonic fibroblasts.J Biol Chem. 2011 May 6;286(18):16030-8. doi: 10.1074/jbc.M110.199398. Epub 2011 Mar 14. J Biol Chem. 2011. PMID: 21402702 Free PMC article.
-
The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity.Cardiovasc Drugs Ther. 2020 Apr;34(2):255-269. doi: 10.1007/s10557-020-06941-x. Cardiovasc Drugs Ther. 2020. PMID: 32034646 Free PMC article. Review.
-
Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy.J Mol Cell Cardiol. 2011 Oct;51(4):614-8. doi: 10.1016/j.yjmcc.2011.01.008. Epub 2011 Jan 27. J Mol Cell Cardiol. 2011. PMID: 21276800 Free PMC article. Review.
Cited by
-
Doxorubicin cardiomyopathy is ameliorated by acacetin via Sirt1-mediated activation of AMPK/Nrf2 signal molecules.J Cell Mol Med. 2020 Oct;24(20):12141-12153. doi: 10.1111/jcmm.15859. Epub 2020 Sep 11. J Cell Mol Med. 2020. PMID: 32918384 Free PMC article.
-
CTRP5-Overexpression Attenuated Ischemia-Reperfusion Associated Heart Injuries and Improved Infarction Induced Heart Failure.Front Pharmacol. 2020 Dec 22;11:603322. doi: 10.3389/fphar.2020.603322. eCollection 2020. Front Pharmacol. 2020. PMID: 33414720 Free PMC article.
-
Nuclear translocation of mitochondrial dehydrogenases as an adaptive cardioprotective mechanism.Nat Commun. 2023 Jul 19;14(1):4360. doi: 10.1038/s41467-023-40084-5. Nat Commun. 2023. PMID: 37468519 Free PMC article.
-
Impact of Cerium Oxide Nanoparticles on Metabolic, Apoptotic, Autophagic and Antioxidant Changes in Doxorubicin-Induced Cardiomyopathy: Possible Underlying Mechanisms.Rep Biochem Mol Biol. 2023 Oct;12(3):495-511. doi: 10.61186/rbmb.12.3.495. Rep Biochem Mol Biol. 2023. PMID: 38618259 Free PMC article.
-
Proteomic profiling and genome-wide mapping of O-GlcNAc chromatin-associated proteins reveal an O-GlcNAc-regulated genotoxic stress response.Nat Commun. 2020 Nov 19;11(1):5898. doi: 10.1038/s41467-020-19579-y. Nat Commun. 2020. PMID: 33214551 Free PMC article.
References
-
- Kalyanaraman B., Joseph J., Kalivendi S., Wang S., Konorev E., Kotamraju S. (2002) Doxorubicin-induced apoptosis implications in cardiotoxicity. Mol. Cell Biochem. 234–235, 119–124 - PubMed
-
- Kang Y. J., Chen Y., Epstein P. N. (1996) Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 271, 12610–12616 - PubMed
-
- Ewer M. S., Ewer S. M. (2010) Cardiotoxicity of anticancer treatments: What the cardiologist needs to know. Nat. Rev. Cardiol. 7, 564–575 - PubMed
-
- Ferreira A. L., Matsubara L. S., Matsubara B. B. (2008) Anthracycline-induced cardiotoxicity. Cardiovasc. Hematol. Agents Med. Chem. 6, 278–281 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL074399/HL/NHLBI NIH HHS/United States
- R01 HL089920/HL/NHLBI NIH HHS/United States
- HL080499/HL/NHLBI NIH HHS/United States
- HL110448/HL/NHLBI NIH HHS/United States
- R01 HL079584/HL/NHLBI NIH HHS/United States
- R44 HL110448/HL/NHLBI NIH HHS/United States
- R43 HL110448/HL/NHLBI NIH HHS/United States
- R01 HL080499/HL/NHLBI NIH HHS/United States
- R01 HL096032/HL/NHLBI NIH HHS/United States
- HL096032/HL/NHLBI NIH HHS/United States
- R01 HL074399/HL/NHLBI NIH HHS/United States
- HL089920/HL/NHLBI NIH HHS/United States
- HL079584/HL/NHLBI NIH HHS/United States
- HL105157/HL/NHLBI NIH HHS/United States
- R01 HL110488/HL/NHLBI NIH HHS/United States
- R01 HL105157/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
