

Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression
- PMID: 22271573
- PMCID: PMC3280870
- DOI: 10.1084/jem.20111117
Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression
Erratum in
- J Exp Med. 2012 Mar 12;209(3):640
Abstract
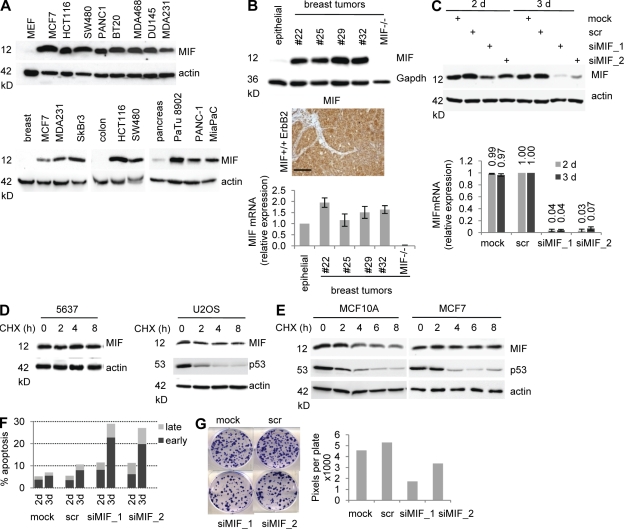
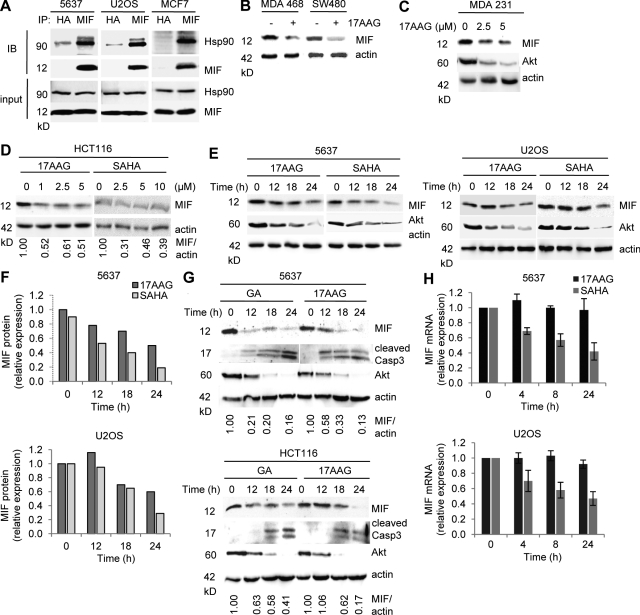
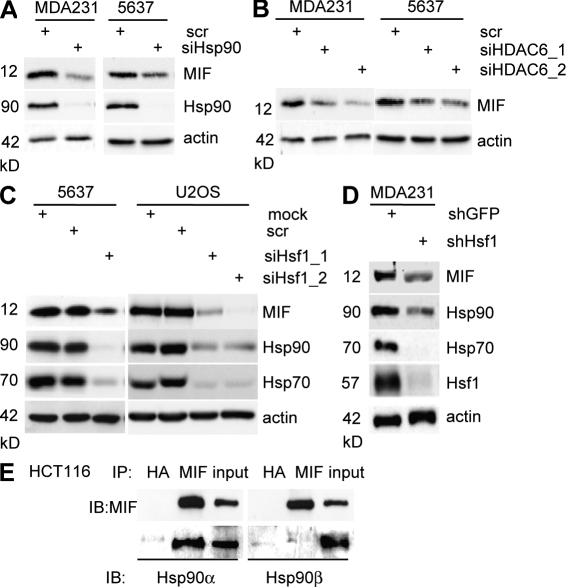
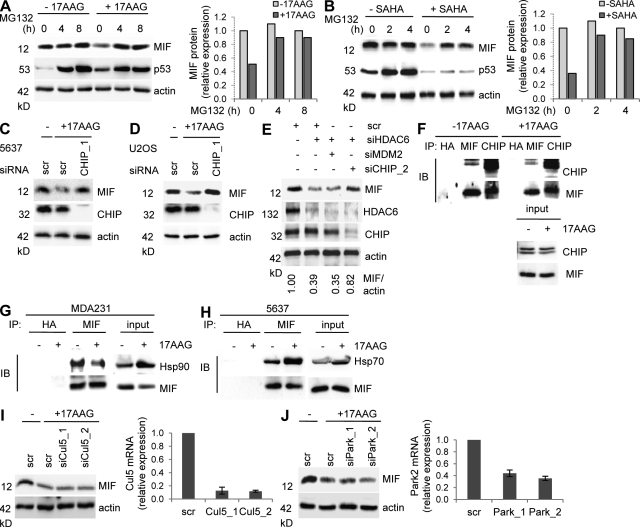
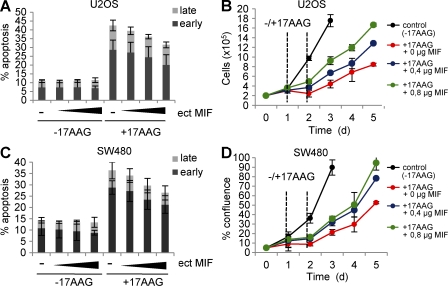
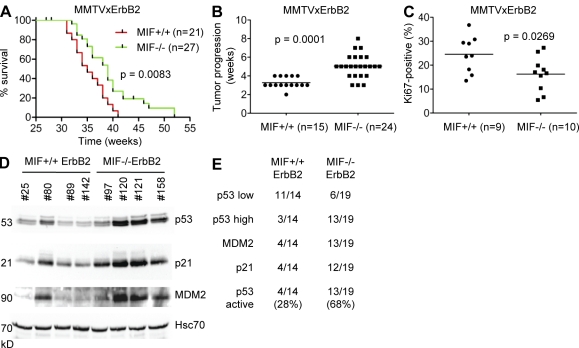
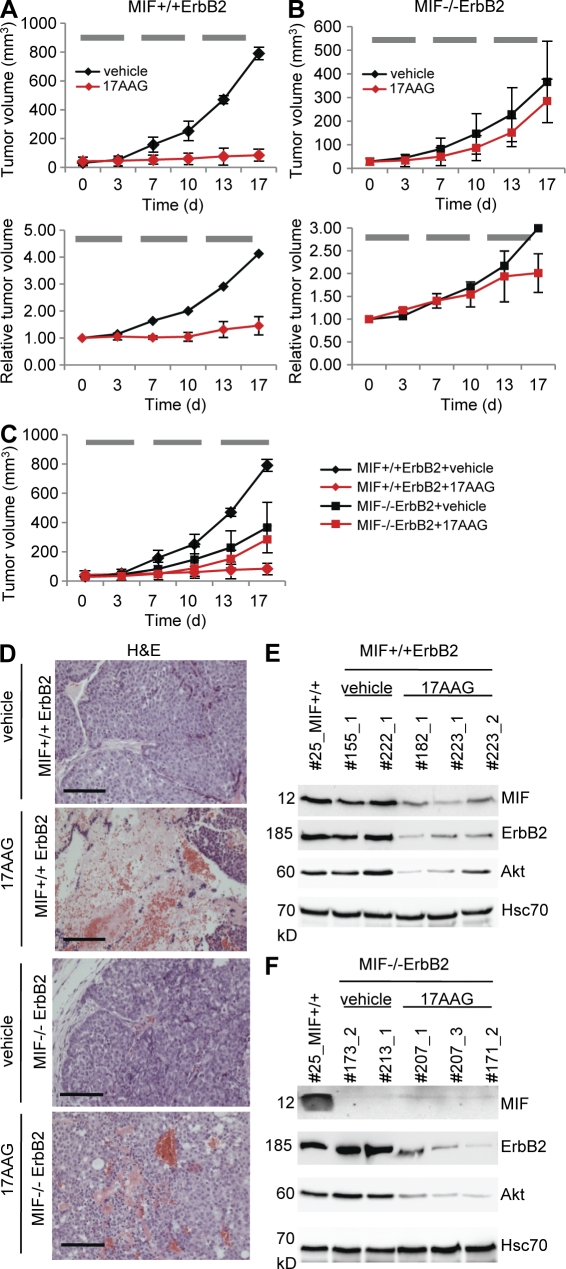
Intracellular macrophage migration inhibitory factor (MIF) often becomes stabilized in human cancer cells. MIF can promote tumor cell survival, and elevated MIF protein correlates with tumor aggressiveness and poor prognosis. However, the molecular mechanism facilitating MIF stabilization in tumors is not understood. We show that the tumor-activated HSP90 chaperone complex protects MIF from degradation. Pharmacological inhibition of HSP90 activity, or siRNA-mediated knockdown of HSP90 or HDAC6, destabilizes MIF in a variety of human cancer cells. The HSP90-associated E3 ubiquitin ligase CHIP mediates the ensuing proteasome-dependent MIF degradation. Cancer cells contain constitutive endogenous MIF-HSP90 complexes. siRNA-mediated MIF knockdown inhibits proliferation and triggers apoptosis of cultured human cancer cells, whereas HSP90 inhibitor-induced apoptosis is overridden by ectopic MIF expression. In the ErbB2 transgenic model of human HER2-positive breast cancer, genetic ablation of MIF delays tumor progression and prolongs overall survival of mice. Systemic treatment with the HSP90 inhibitor 17AAG reduces MIF expression and blocks growth of MIF-expressing, but not MIF-deficient, tumors. Together, these findings identify MIF as a novel HSP90 client and suggest that HSP90 inhibitors inhibit ErbB2-driven breast tumor growth at least in part by destabilizing MIF.
Figures
Similar articles
-
HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer.Cell Death Dis. 2014 Jan 2;5(1):e980. doi: 10.1038/cddis.2013.508. Cell Death Dis. 2014. PMID: 24384723 Free PMC article.
-
E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells.Proc Natl Acad Sci U S A. 2014 May 6;111(18):6834-9. doi: 10.1073/pnas.1322412111. Epub 2014 Apr 23. Proc Natl Acad Sci U S A. 2014. PMID: 24760825 Free PMC article.
-
Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer.Cell Mol Biol Lett. 2012 Sep;17(3):446-58. doi: 10.2478/s11658-012-0021-8. Epub 2012 Jun 5. Cell Mol Biol Lett. 2012. PMID: 22669480 Free PMC article.
-
Targeting the heat shock protein 90: a rational way to inhibit macrophage migration inhibitory factor function in cancer.Curr Opin Oncol. 2014 Jan;26(1):108-13. doi: 10.1097/CCO.0000000000000036. Curr Opin Oncol. 2014. PMID: 24225413 Review.
-
Macrophage migration inhibitory factor manipulation and evaluation in tumoral hypoxic adaptation.Methods Enzymol. 2007;435:355-69. doi: 10.1016/S0076-6879(07)35018-0. Methods Enzymol. 2007. PMID: 17998063 Free PMC article. Review.
Cited by
-
Small interfering RNA (siRNA)-mediated knockdown of macrophage migration inhibitory factor (MIF) suppressed cyclin D1 expression and hepatocellular carcinoma cell proliferation.Oncotarget. 2014 Jul 30;5(14):5570-80. doi: 10.18632/oncotarget.2141. Oncotarget. 2014. PMID: 25015194 Free PMC article.
-
Involvement of macrophage migration inhibitory factor and its receptor (CD74) in human breast cancer.Oncol Rep. 2014 Aug;32(2):523-9. doi: 10.3892/or.2014.3272. Epub 2014 Jun 16. Oncol Rep. 2014. PMID: 24939415 Free PMC article.
-
HSP90 inhibitor antagonizing MIF: The specifics of pleiotropic cancer drug candidates.Oncoimmunology. 2012 Nov 1;1(8):1425-1426. doi: 10.4161/onci.21173. Oncoimmunology. 2012. PMID: 23243616 Free PMC article.
-
Destabilization of macrophage migration inhibitory factor by 4-IPP reduces NF-κB/P-TEFb complex-mediated c-Myb transcription to suppress osteosarcoma tumourigenesis.Clin Transl Med. 2022 Jan;12(1):e652. doi: 10.1002/ctm2.652. Clin Transl Med. 2022. PMID: 35060345 Free PMC article.
-
HDAC6 controls innate immune and autophagy responses to TLR-mediated signalling by the intracellular bacteria Listeria monocytogenes.PLoS Pathog. 2017 Dec 27;13(12):e1006799. doi: 10.1371/journal.ppat.1006799. eCollection 2017 Dec. PLoS Pathog. 2017. PMID: 29281743 Free PMC article.
References
-
- Bali P., Pranpat M., Bradner J., Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja P., et al. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280:26729–26734 10.1074/jbc.C500186200 - DOI - PubMed
-
- Banerji U., Walton M., Raynaud F., Grimshaw R., Kelland L., Valenti M., Judson I., Workman P. 2005. Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin. Cancer Res. 11:7023–7032 10.1158/1078-0432.CCR-05-0518 - DOI - PubMed
-
- Barrington R.E., Subler M.A., Rands E., Omer C.A., Miller P.J., Hundley J.E., Koester S.K., Troyer D.A., Bearss D.J., Conner M.W., et al. 1998. A farnesyltransferase inhibitor induces tumor regression in transgenic mice harboring multiple oncogenic mutations by mediating alterations in both cell cycle control and apoptosis. Mol. Cell. Biol. 18:85–92 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
