

Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion
- PMID: 22271886
- PMCID: PMC3396667
- DOI: 10.3324/haematol.2011.053504
Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion
Abstract
Background: The clinical and hematologic features of β-thalassemia are modulated by different factors, resulting in a wide range of clinical severity. The main factors are the type of disease-causing mutation and the ability to produce α-globin and γ-globin chains. In the present study we investigated the respective contributions of known modifiers to the prediction of the clinical severity of β-thalassemia as assessed by the patients' age at first transfusion.
Design and methods: We studied the effect of seven loci in a cohort of 316 Sardinian patients with β(0)-thalassemia. In addition to characterizing the β-globin gene mutations, α-globin gene defects and HBG2:g.-158C>T polymorphism, we genotyped two different markers in the BCL11A gene and three in the HBS1L-MYB intergenic region using single nucleotide polymorphism microarrays, imputation and direct genotyping. We performed Cox proportional hazard analysis of the time to first transfusion.
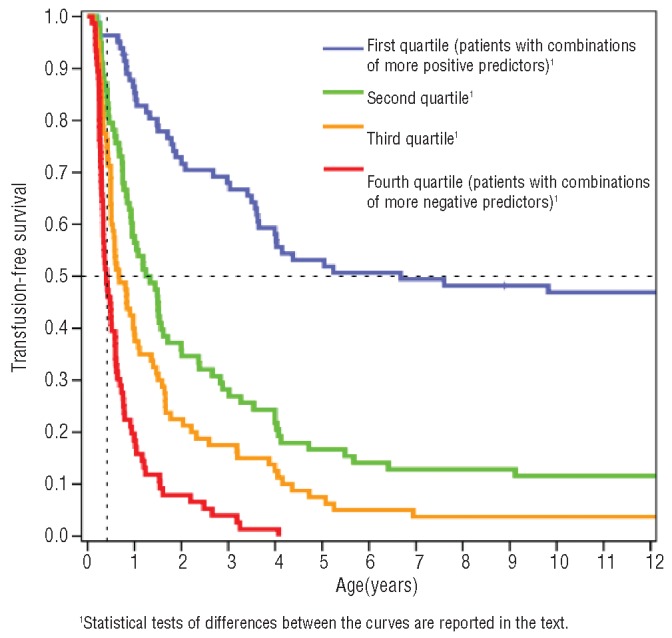
Results: According to the resulting model, we were able to explain phenotypic severity to a large extent (Harrell's concordance index=0.72; Cox & Snell R(2)=0.394) and demonstrated that most of the model's discriminatory ability is attributable to the genetic variants affecting fetal hemoglobin production (HBG2:g.-158C>T, BCL11A and HBS1L-MYB loci: C-index=0.68, R(2)=0.272), while the remaining is due to α-globin gene defects and gender. Consequently, significantly distinct survival curves can be described in our population.
Conclusions: This detailed analysis clarifies the impact of genetic modifiers on the clinical severity of the disease, measured by time to first transfusion, by determining their relative contributions in a homogeneous cohort of β(0)-thalassemia patients. It may also support clinical decisions regarding the beginning of transfusion therapy in patients with β-thalassemia.
Figures
Similar articles
-
A genetic score for the prediction of beta-thalassemia severity.Haematologica. 2015 Apr;100(4):452-7. doi: 10.3324/haematol.2014.113886. Epub 2014 Dec 5. Haematologica. 2015. PMID: 25480500 Free PMC article.
-
Existence of HbF Enhancer Haplotypes at HBS1L-MYB Intergenic Region in Transfusion-Dependent Saudi β-Thalassemia Patients.Biomed Res Int. 2017;2017:1972429. doi: 10.1155/2017/1972429. Epub 2017 Feb 9. Biomed Res Int. 2017. PMID: 28280727 Free PMC article.
-
Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: A study on β-thalassemia and hemoglobin E/β-thalassemia patients in Indonesia.Hematol Oncol Stem Cell Ther. 2016 Jun;9(2):55-63. doi: 10.1016/j.hemonc.2016.02.003. Epub 2016 Mar 17. Hematol Oncol Stem Cell Ther. 2016. PMID: 27009595
-
Genetic Basis and Genetic Modifiers of β-Thalassemia and Sickle Cell Disease.Adv Exp Med Biol. 2017;1013:27-57. doi: 10.1007/978-1-4939-7299-9_2. Adv Exp Med Biol. 2017. PMID: 29127676 Review.
-
Hemoglobin switching's surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin.Curr Opin Genet Dev. 2015 Aug;33:62-70. doi: 10.1016/j.gde.2015.08.001. Epub 2015 Sep 14. Curr Opin Genet Dev. 2015. PMID: 26375765 Free PMC article. Review.
Cited by
-
BCL11A Polymorphism in Egyptian Children with β-Thalassemia: Relation to Phenotypic Heterogeneity.J Pediatr Genet. 2021 Jun 1;12(1):16-22. doi: 10.1055/s-0041-1728744. eCollection 2023 Mar. J Pediatr Genet. 2021. PMID: 36684548 Free PMC article.
-
HBB mutations and HbA2 level: Escaping the carrier screening programs.Clin Case Rep. 2020 Dec 29;9(2):973-977. doi: 10.1002/ccr3.3714. eCollection 2021 Feb. Clin Case Rep. 2020. PMID: 33598281 Free PMC article.
-
Genotyping of BCL11A and HBS1L-MYB SNPs associated with fetal haemoglobin levels: a SNaPshot minisequencing approach.BMC Genomics. 2014 Feb 6;15:108. doi: 10.1186/1471-2164-15-108. BMC Genomics. 2014. PMID: 24502199 Free PMC article.
-
Pharmacogenomics of Drugs Used in β-Thalassemia and Sickle-Cell Disease: From Basic Research to Clinical Applications.Int J Mol Sci. 2024 Apr 12;25(8):4263. doi: 10.3390/ijms25084263. Int J Mol Sci. 2024. PMID: 38673849 Free PMC article. Review.
-
Non-transfusion-dependent thalassemias.Haematologica. 2013 Jun;98(6):833-44. doi: 10.3324/haematol.2012.066845. Haematologica. 2013. PMID: 23729725 Free PMC article. Review.
References
-
- Cao A, Galanello R. Beta-Thalassemia. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993–2000. [updated 2010]
-
- Cao A, Rosatelli C, Pirastu M, Galanello R. Thalassemias in Sardinia: molecular pathology, phenotype-genotype correlation, and prevention. Am J Pediatr Hematol Oncol. 1991;13(2):179–88. - PubMed
-
- Cao A, Galanello R, Rosatelli MC. Genotype-phenotype correlations in beta-thalassemias. Blood Rev. 1994;8(1):1–12. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
