

Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates
- PMID: 22272183
- PMCID: PMC3260318
- DOI: 10.1371/journal.pbio.1001241
Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates
Abstract
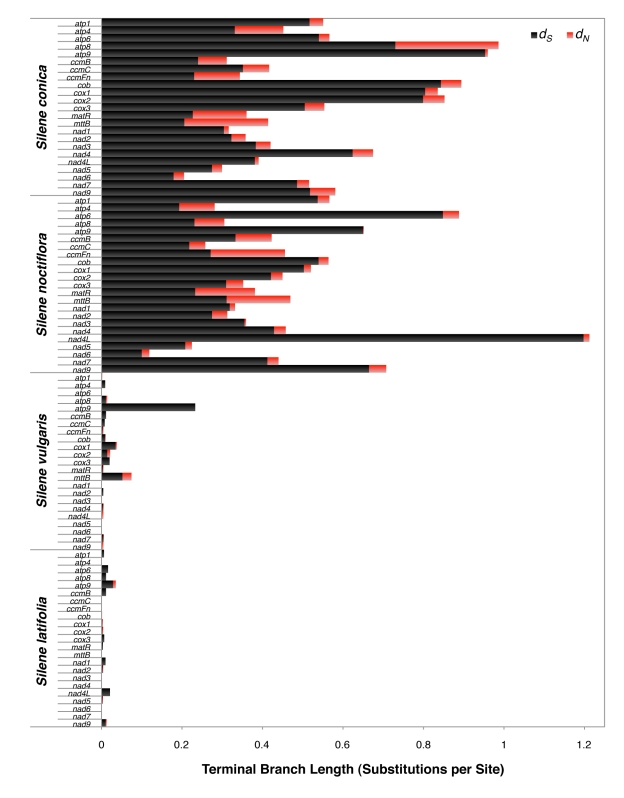
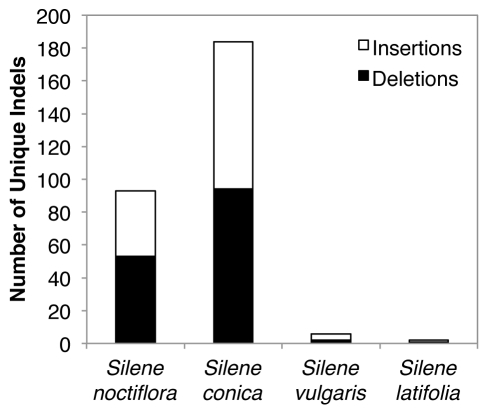
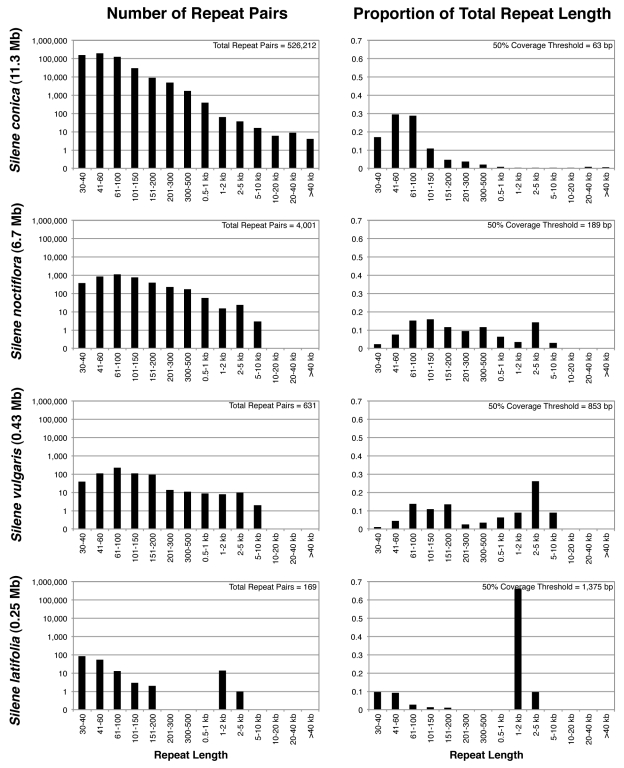
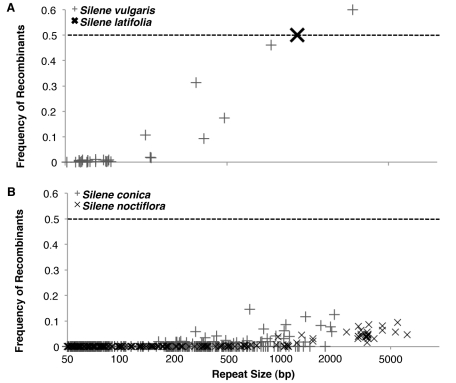
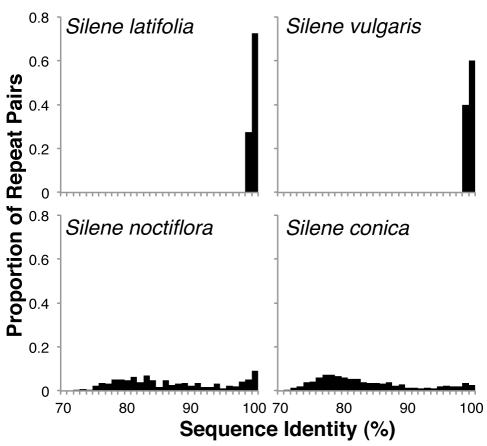
Genome size and complexity vary tremendously among eukaryotic species and their organelles. Comparisons across deeply divergent eukaryotic lineages have suggested that variation in mutation rates may explain this diversity, with increased mutational burdens favoring reduced genome size and complexity. The discovery that mitochondrial mutation rates can differ by orders of magnitude among closely related angiosperm species presents a unique opportunity to test this hypothesis. We sequenced the mitochondrial genomes from two species in the angiosperm genus Silene with recent and dramatic accelerations in their mitochondrial mutation rates. Contrary to theoretical predictions, these genomes have experienced a massive proliferation of noncoding content. At 6.7 and 11.3 Mb, they are by far the largest known mitochondrial genomes, larger than most bacterial genomes and even some nuclear genomes. In contrast, two slowly evolving Silene mitochondrial genomes are smaller than average for angiosperms. Consequently, this genus captures approximately 98% of known variation in organelle genome size. The expanded genomes reveal several architectural changes, including the evolution of complex multichromosomal structures (with 59 and 128 circular-mapping chromosomes, ranging in size from 44 to 192 kb). They also exhibit a substantial reduction in recombination and gene conversion activity as measured by the relative frequency of alternative genome conformations and the level of sequence divergence between repeat copies. The evolution of mutation rate, genome size, and chromosome structure can therefore be extremely rapid and interrelated in ways not predicted by current evolutionary theories. Our results raise the hypothesis that changes in recombinational processes, including gene conversion, may be a central force driving the evolution of both mutation rate and genome structure.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Mira A, Ochman H, Moran N. A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. - PubMed
-
- Petrov D. A. Mutational equilibrium model of genome size evolution. Theor Popul Biol. 2002;61:531–544. - PubMed
-
- Orgel L. E, Crick F. H. Selfish DNA: the ultimate parasite. Nature. 1980;284:604–607. - PubMed
-
- Doolittle W. F, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284:601–603. - PubMed
-
- Cavalier-Smith T. Skeletal DNA and the evolution of genome size. Annu Rev Biophys Bioeng. 1982;11:273–302. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
