

Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1
- PMID: 22275899
- PMCID: PMC3254186
- DOI: 10.3389/fphar.2011.00075
Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1
Abstract
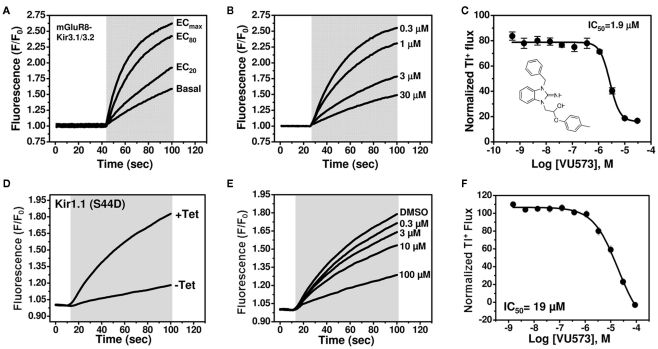
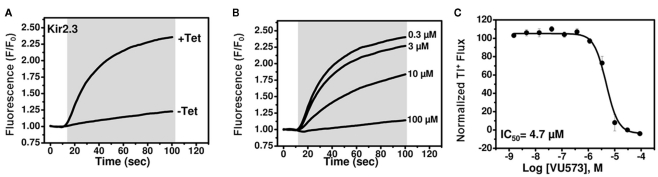
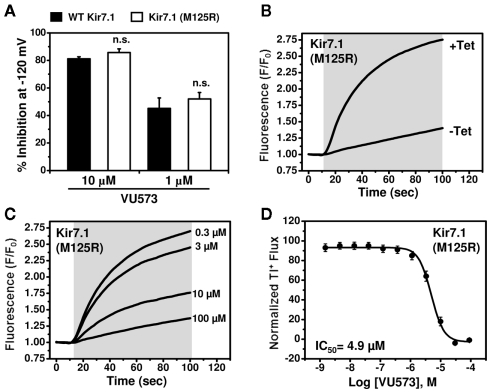
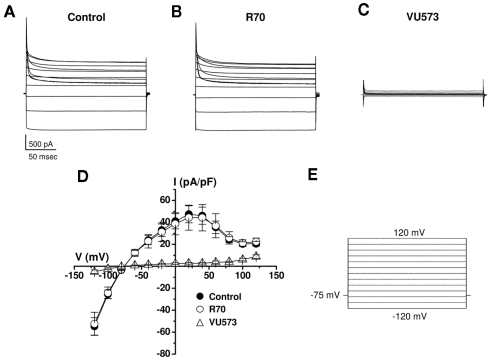
The inward rectifier family of potassium (Kir) channels is comprised of at least 16 family members exhibiting broad and often overlapping cellular, tissue, or organ distributions. The discovery of disease-causing mutations in humans and experiments on knockout mice has underscored the importance of Kir channels in physiology and in some cases raised questions about their potential as drug targets. However, the paucity of potent and selective small-molecule modulators targeting specific family members has with few exceptions mired efforts to understand their physiology and assess their therapeutic potential. A growing body of evidence suggests that G protein-coupled inward rectifier K (GIRK) channels of the Kir3.X subfamily may represent novel targets for the treatment of atrial fibrillation. In an effort to expand the molecular pharmacology of GIRK, we performed a thallium (Tl(+)) flux-based high-throughput screen of a Kir1.1 inhibitor library for modulators of GIRK. One compound, termed VU573, exhibited 10-fold selectivity for GIRK over Kir1.1 (IC(50) = 1.9 and 19 μM, respectively) and was therefore selected for further study. In electrophysiological experiments performed on Xenopus laevis oocytes and mammalian cells, VU573 inhibited Kir3.1/3.2 (neuronal GIRK) and Kir3.1/3.4 (cardiac GIRK) channels with equal potency and preferentially inhibited GIRK, Kir2.3, and Kir7.1 over Kir1.1 and Kir2.1.Tl(+) flux assays were established for Kir2.3 and the M125R pore mutant of Kir7.1 to support medicinal chemistry efforts to develop more potent and selective analogs for these channels. The structure-activity relationships of VU573 revealed few analogs with improved potency, however two compounds retained most of their activity toward GIRK and Kir2.3 and lost activity toward Kir7.1. We anticipate that the VU573 series will be useful for exploring the physiology and structure-function relationships of these Kir channels.
Keywords: GIRK; electrophysiology; fluorescence; high throughput; pharmacology; screening; thallium flux.
Figures
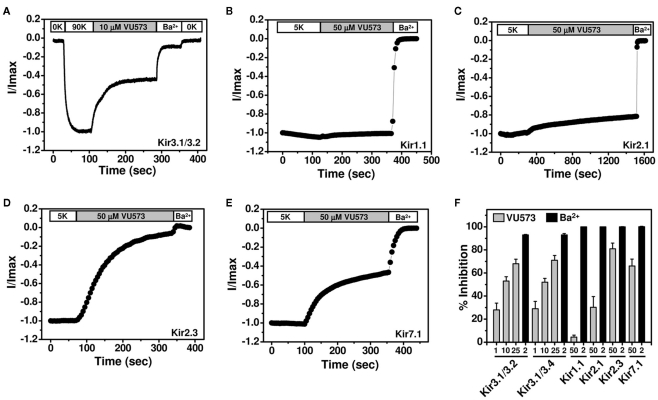
 ) or Ba2+ (■) n = 4–6).
) or Ba2+ (■) n = 4–6).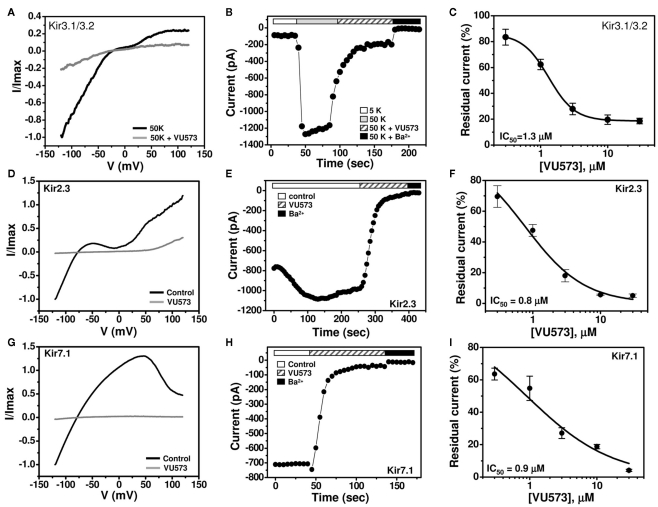
Comment in
-
Finding inward rectifier channel inhibitors: why and how?Front Pharmacol. 2012 Jan 9;2:95. doi: 10.3389/fphar.2011.00095. eCollection 2011. Front Pharmacol. 2012. PMID: 22291650 Free PMC article. No abstract available.
Similar articles
-
Screening Technologies for Inward Rectifier Potassium Channels: Discovery of New Blockers and Activators.SLAS Discov. 2020 Jun;25(5):420-433. doi: 10.1177/2472555220905558. Epub 2020 Apr 15. SLAS Discov. 2020. PMID: 32292089
-
ML418: The First Selective, Sub-Micromolar Pore Blocker of Kir7.1 Potassium Channels.ACS Chem Neurosci. 2016 Jul 20;7(7):1013-23. doi: 10.1021/acschemneuro.6b00111. Epub 2016 May 24. ACS Chem Neurosci. 2016. PMID: 27184474 Free PMC article.
-
High-throughput screening for small-molecule modulators of inward rectifier potassium channels.J Vis Exp. 2013 Jan 27;(71):4209. doi: 10.3791/4209. J Vis Exp. 2013. PMID: 23381507 Free PMC article.
-
Next-generation inward rectifier potassium channel modulators: discovery and molecular pharmacology.Am J Physiol Cell Physiol. 2021 Jun 1;320(6):C1125-C1140. doi: 10.1152/ajpcell.00548.2020. Epub 2021 Apr 7. Am J Physiol Cell Physiol. 2021. PMID: 33826405 Free PMC article. Review.
-
Discovery of a small molecule inhibitor of ROMK and Kir7.1.2009 Sep 1 [updated 2010 Oct 20]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010–. 2009 Sep 1 [updated 2010 Oct 20]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010–. PMID: 21433378 Free Books & Documents. Review.
Cited by
-
Development and validation of fluorescence-based and automated patch clamp-based functional assays for the inward rectifier potassium channel Kir4.1.Assay Drug Dev Technol. 2013 Nov-Dec;11(9-10):532-43. doi: 10.1089/adt.2013.544. Epub 2013 Nov 22. Assay Drug Dev Technol. 2013. PMID: 24266659 Free PMC article.
-
Novel Fluorescence-Based High-Throughput FLIPR Assay Utilizing Membrane-Tethered Genetic Calcium Sensors to Identify T-Type Calcium Channel Modulators.ACS Pharmacol Transl Sci. 2022 Feb 25;5(3):156-168. doi: 10.1021/acsptsci.1c00233. eCollection 2022 Mar 11. ACS Pharmacol Transl Sci. 2022. PMID: 35311021 Free PMC article.
-
Novel KCNJ10 Gene Variations Compromise Function of Inwardly Rectifying Potassium Channel 4.1.J Biol Chem. 2016 Apr 1;291(14):7716-26. doi: 10.1074/jbc.M115.679910. Epub 2016 Feb 11. J Biol Chem. 2016. PMID: 26867573 Free PMC article.
-
Role and mechanisms of regulation of the basolateral Kir 4.1/Kir 5.1K+ channels in the distal tubules.Acta Physiol (Oxf). 2017 Jan;219(1):260-273. doi: 10.1111/apha.12703. Epub 2016 May 20. Acta Physiol (Oxf). 2017. PMID: 27129733 Free PMC article. Review.
-
Phylogenomics of Tick Inward Rectifier Potassium Channels and Their Potential as Targets to Innovate Control Technologies.Front Cell Infect Microbiol. 2021 Mar 19;11:647020. doi: 10.3389/fcimb.2021.647020. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 33816352 Free PMC article. Review.
References
-
- Bhave G., Chauder B. A., Liu W., Dawson E. S., Kadakia R., Nguyen T. T., Lewis L. M., Meiler J., Weaver C. D., Satlin L. M., Lindsley C. W., Denton J. S. (2011). Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol. Pharmacol. 79, 42–5010.1124/mol.110.066928 - DOI - PMC - PubMed
-
- Caballero R., Dolz-Gaiton P., Gomez R., Amoros I., Barana A., Gonzalez De La Fuente M., Osuna L., Duarte J., Lopez-Izquierdo A., Moraleda I., Galvez E., Sanchez-Chapula J. A., Tamargo J., Delpon E. (2010). Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. U.S.A. 107, 15631–1563610.1073/pnas.1004021107 - DOI - PMC - PubMed
-
- Caroti P., Ceccotti C., Da Settimo A., Palla F., Primofiore G. (1986). A facile synthesis of 5,7-dihydro-5-oxopyrido[3′,2′:5,6]pyrimido[1,2-a]benzimidazoles. A new heterocyclic ring system. J. Heterocycl. Chem. 82, 1833–183610.1002/jhet.5570230645 - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases