

Fast computation and applications of genome mappability
- PMID: 22276185
- PMCID: PMC3261895
- DOI: 10.1371/journal.pone.0030377
Fast computation and applications of genome mappability
Abstract
We present a fast mapping-based algorithm to compute the mappability of each region of a reference genome up to a specified number of mismatches. Knowing the mappability of a genome is crucial for the interpretation of massively parallel sequencing experiments. We investigate the properties of the mappability of eukaryotic DNA/RNA both as a whole and at the level of the gene family, providing for various organisms tracks which allow the mappability information to be visually explored. In addition, we show that mappability varies greatly between species and gene classes. Finally, we suggest several practical applications where mappability can be used to refine the analysis of high-throughput sequencing data (SNP calling, gene expression quantification and paired-end experiments). This work highlights mappability as an important concept which deserves to be taken into full account, in particular when massively parallel sequencing technologies are employed. The GEM mappability program belongs to the GEM (GEnome Multitool) suite of programs, which can be freely downloaded for any use from its website (http://gemlibrary.sourceforge.net).
Conflict of interest statement
Figures
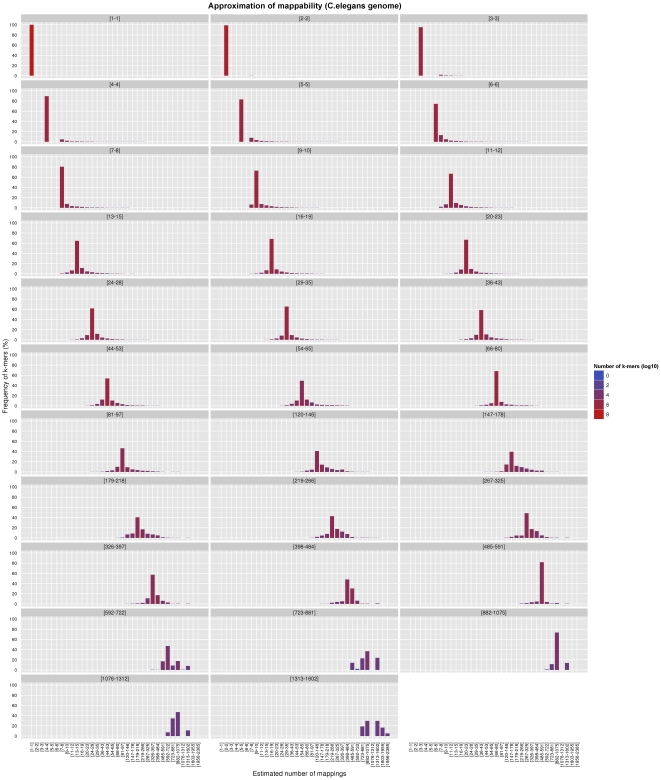
 and
and
 . Both the exact and the approximated data were obtained with gem-mappability, the former by setting the value of parameter
. Both the exact and the approximated data were obtained with gem-mappability, the former by setting the value of parameter  to
to  , the latter with the default value of
, the latter with the default value of  automatically selected by the program after the length of the C.elegans genome. Each panel shows how our approximation scatters the
automatically selected by the program after the length of the C.elegans genome. Each panel shows how our approximation scatters the  -mers originally populating a non-approximate
-mers originally populating a non-approximate  -bit frequence bin into more than one single approximate bin. Using the panel – as an example, one can see that about 80% of the
-bit frequence bin into more than one single approximate bin. Using the panel – as an example, one can see that about 80% of the  -mers fall into the correct bin, while the remaining 20% is dispersed in bins from – to –, with most of the
-mers fall into the correct bin, while the remaining 20% is dispersed in bins from – to –, with most of the  -mers staying in bins close to the correct one. In addition, the color of the bins shows that such a 20% of
-mers staying in bins close to the correct one. In addition, the color of the bins shows that such a 20% of  -mers corresponds in absolute terms to a small number (in this example about the 90% of the
-mers corresponds in absolute terms to a small number (in this example about the 90% of the  -mers of the genome is unique and hence falls into the [1–1] bin, which, as explained in the text, is not perturbed by our approximation owing to the good properties of the latter).
-mers of the genome is unique and hence falls into the [1–1] bin, which, as explained in the text, is not perturbed by our approximation owing to the good properties of the latter).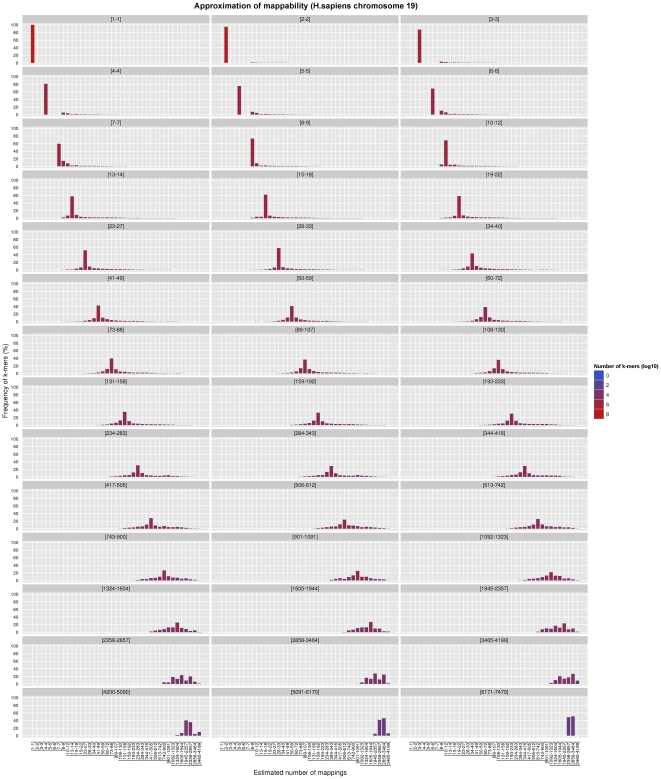
 of
H.sapiens
, for
of
H.sapiens
, for
 and
and
 . Both the exact and the approximated data were obtained with gem-mappability, the former by setting the value of parameter
. Both the exact and the approximated data were obtained with gem-mappability, the former by setting the value of parameter  to
to  , the latter with the default value of
, the latter with the default value of  automatically selected by the program after the length of chromosome
automatically selected by the program after the length of chromosome  of H.sapiens. Each panel shows how our approximation scatters the
of H.sapiens. Each panel shows how our approximation scatters the  -mers originally populating a non-approximate
-mers originally populating a non-approximate  -bit frequence bin into more than one single approximate bin.
-bit frequence bin into more than one single approximate bin.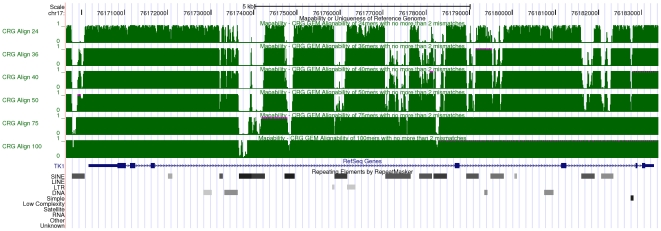
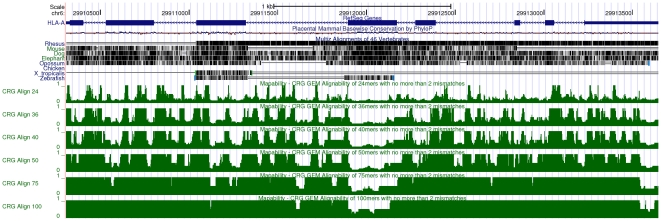
 -mer sizes
-mer sizes  ,
,  ,
,  ,
,  and
and  bp (from top to bottom of the figure). Regions with low mappability score have high frequencies, and conversely. This example illustrates that the uniqueness of the TK1 locus (especially within the introns) could be inversely correlated with the presence of some repetitive elements as identified by RepeatMasker .
bp (from top to bottom of the figure). Regions with low mappability score have high frequencies, and conversely. This example illustrates that the uniqueness of the TK1 locus (especially within the introns) could be inversely correlated with the presence of some repetitive elements as identified by RepeatMasker .
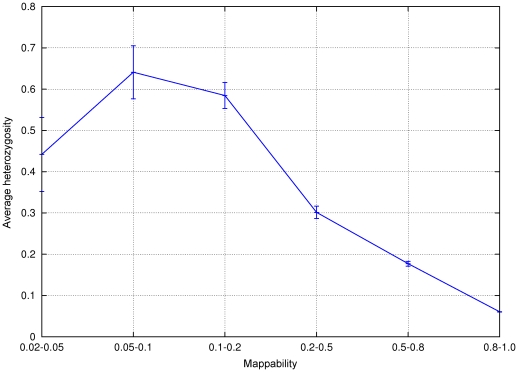
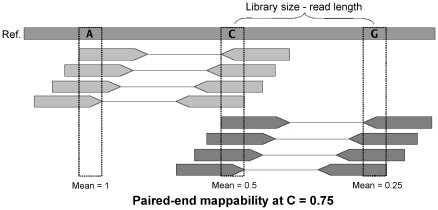
 -mers covering a particular position of the genome (corresponding to nucleotide C) is equal to
-mers covering a particular position of the genome (corresponding to nucleotide C) is equal to  (
( in this example). The average of the mappabilities of the
in this example). The average of the mappabilities of the  -mers can be taken as the pileup mappability. Such a quantity represents how mappable would be this position in a pileup of a whole genome sequencing study with reads of length
-mers can be taken as the pileup mappability. Such a quantity represents how mappable would be this position in a pileup of a whole genome sequencing study with reads of length  .
.
 and
and  out of an in-house experiment with average coverage 30
out of an in-house experiment with average coverage 30 .
.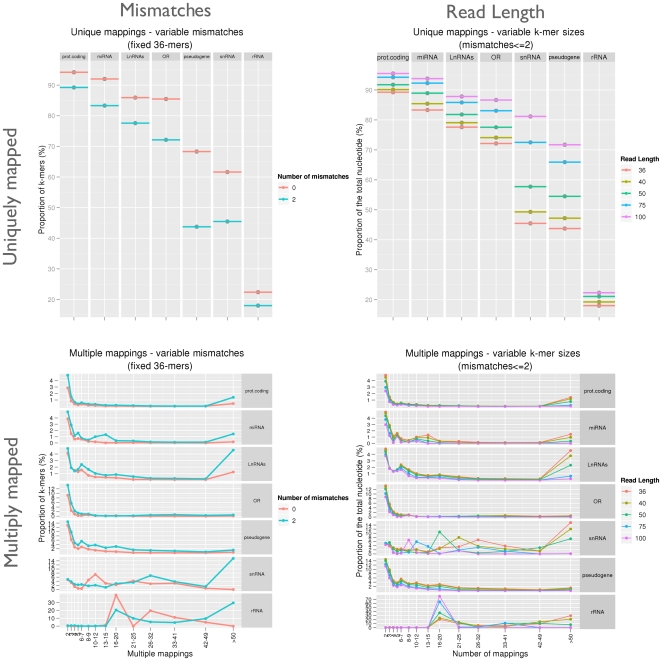
 -mers having a frequency of
-mers having a frequency of  (i.e. uniquely mappable) and those having a frequency
(i.e. uniquely mappable) and those having a frequency  (ambiguous) on the first and second row, respectively. The influence of mismatch number and
(ambiguous) on the first and second row, respectively. The influence of mismatch number and  -mer lengths are presented in the first and second column, respectively.
-mer lengths are presented in the first and second column, respectively.
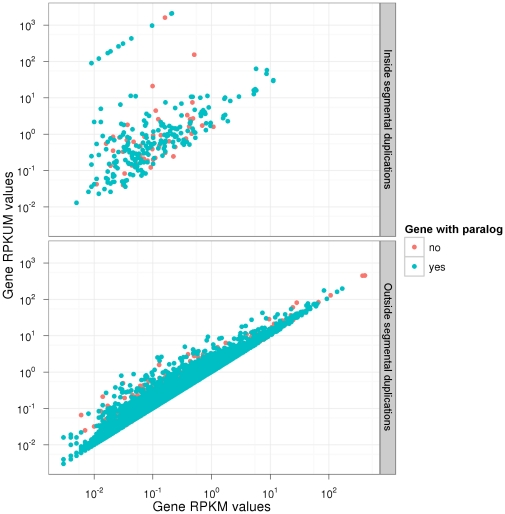
 -
- .
.
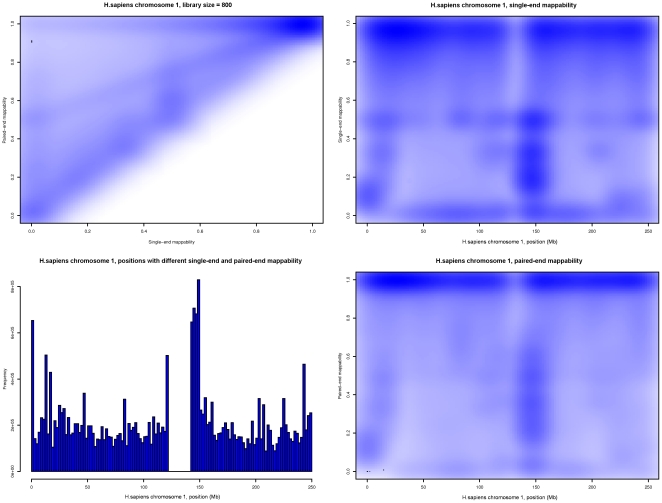
 -mer length 100, 2 mismatches and a library size of 800 bases. Top left: Heatmap of the number of locations in HSA1 as a function of their single-end and paired-end mappabilities. Bottom left: Histogram of the number of locations in HSA1 that show different single-end and paired-end mappabilities, plotted versus their position along the chromosome. Top right: Heatmap of the number of locations in HSA1 as a function of their single-end mappability and their position along the chromosome. Bottom right: Heatmap of the number of locations in HSA1 as a function of their paired-end mappability and their position along the chromosome.
-mer length 100, 2 mismatches and a library size of 800 bases. Top left: Heatmap of the number of locations in HSA1 as a function of their single-end and paired-end mappabilities. Bottom left: Histogram of the number of locations in HSA1 that show different single-end and paired-end mappabilities, plotted versus their position along the chromosome. Top right: Heatmap of the number of locations in HSA1 as a function of their single-end mappability and their position along the chromosome. Bottom right: Heatmap of the number of locations in HSA1 as a function of their paired-end mappability and their position along the chromosome.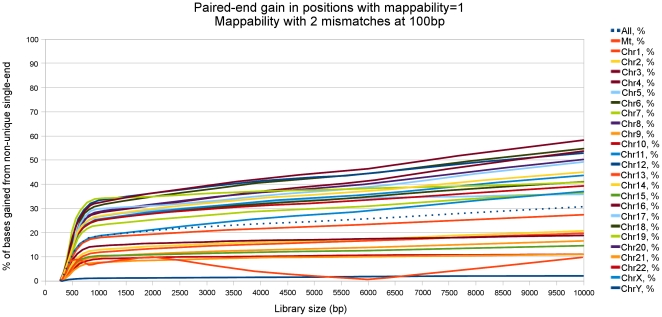
References
-
- Ribeca P. The GEM (GEnome Multitool) library. 2008 URL http://gemlibrary.sourceforge.net. Accessed 2011 Dec 23.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
