

Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases
- PMID: 22277662
- PMCID: PMC3352121
- DOI: 10.1186/1750-1172-7-8
Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases
Abstract
Background: Inherited retinal disorders are clinically and genetically heterogeneous with more than 150 gene defects accounting for the diversity of disease phenotypes. So far, mutation detection was mainly performed by APEX technology and direct Sanger sequencing of known genes. However, these methods are time consuming, expensive and unable to provide a result if the patient carries a new gene mutation. In addition, multiplicity of phenotypes associated with the same gene defect may be overlooked.
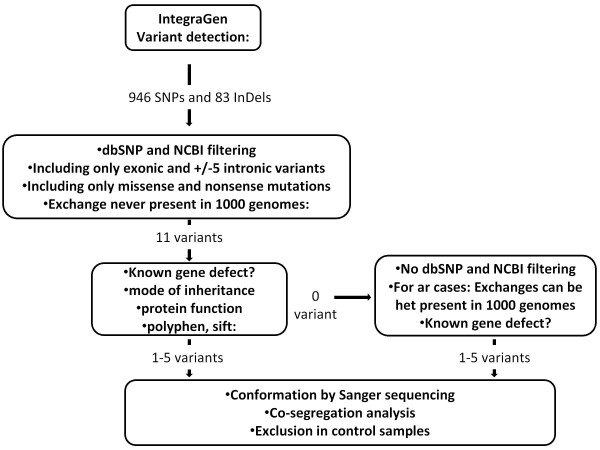
Methods: To overcome these challenges, we designed an exon sequencing array to target 254 known and candidate genes using Agilent capture. Subsequently, 20 DNA samples from 17 different families, including four patients with known mutations were sequenced using Illumina Genome Analyzer IIx next-generation-sequencing (NGS) platform. Different filtering approaches were applied to identify the genetic defect. The most likely disease causing variants were analyzed by Sanger sequencing. Co-segregation and sequencing analysis of control samples validated the pathogenicity of the observed variants.
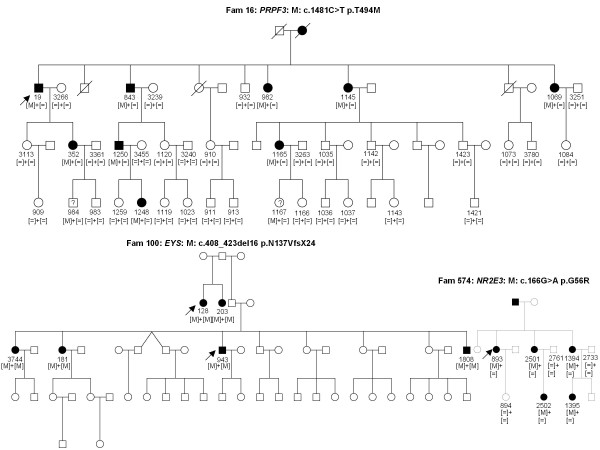
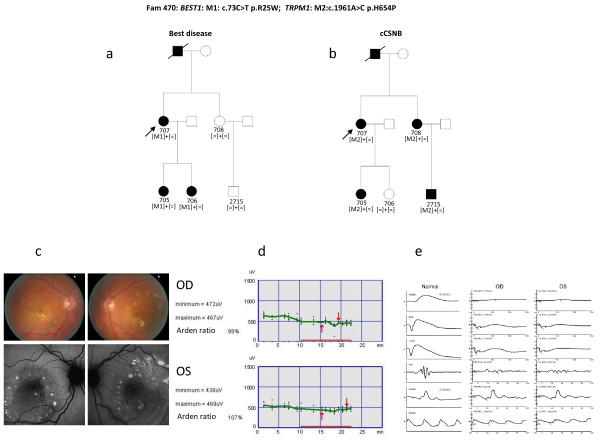
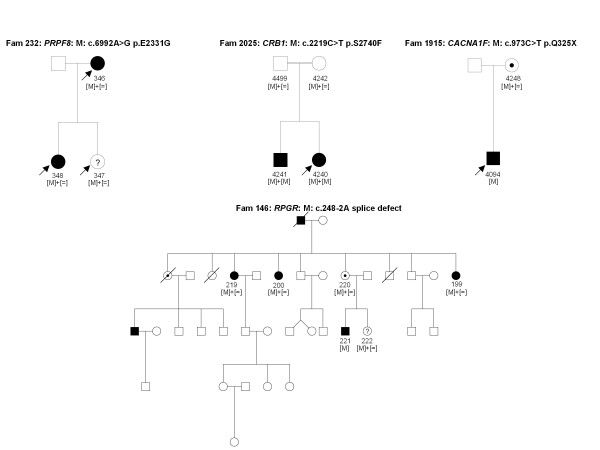
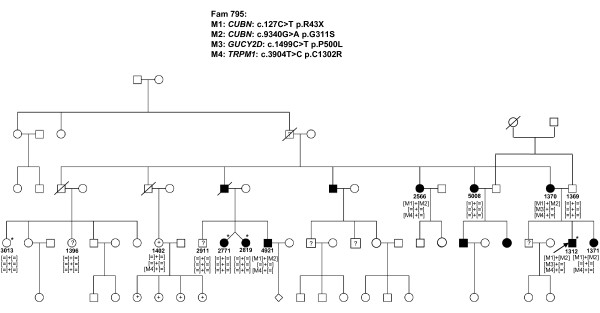
Results: The phenotype of the patients included retinitis pigmentosa, congenital stationary night blindness, Best disease, early-onset cone dystrophy and Stargardt disease. In three of four control samples with known genotypes NGS detected the expected mutations. Three known and five novel mutations were identified in NR2E3, PRPF3, EYS, PRPF8, CRB1, TRPM1 and CACNA1F. One of the control samples with a known genotype belongs to a family with two clinical phenotypes (Best and CSNB), where a novel mutation was identified for CSNB. In six families the disease associated mutations were not found, indicating that novel gene defects remain to be identified.
Conclusions: In summary, this unbiased and time-efficient NGS approach allowed mutation detection in 75% of control cases and in 57% of test cases. Furthermore, it has the possibility of associating known gene defects with novel phenotypes and mode of inheritance.
Figures
References
-
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. - DOI - PMC - PubMed
-
- Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
