

Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins
- PMID: 22278370
- PMCID: PMC3316730
- DOI: 10.1074/mcp.M111.014050
Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins
Abstract
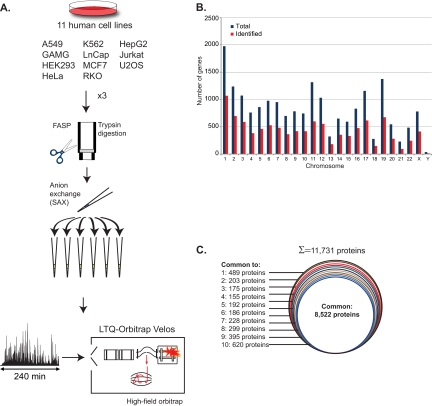
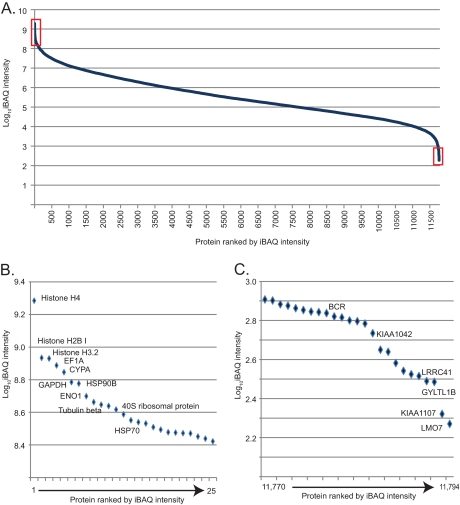
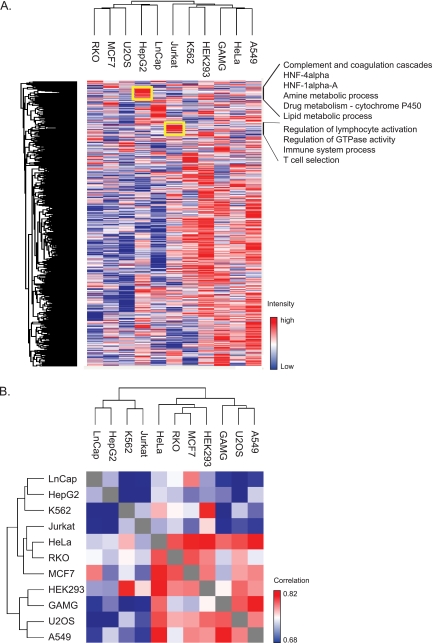
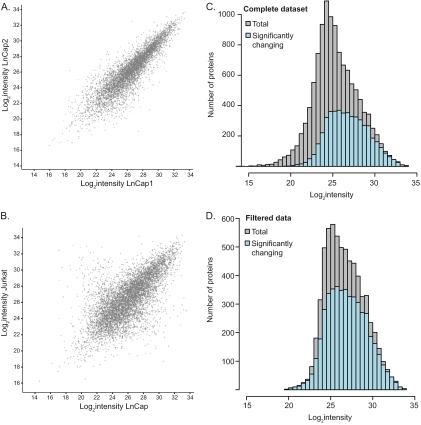
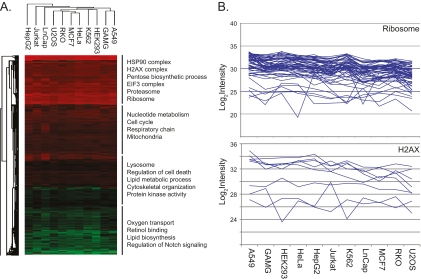
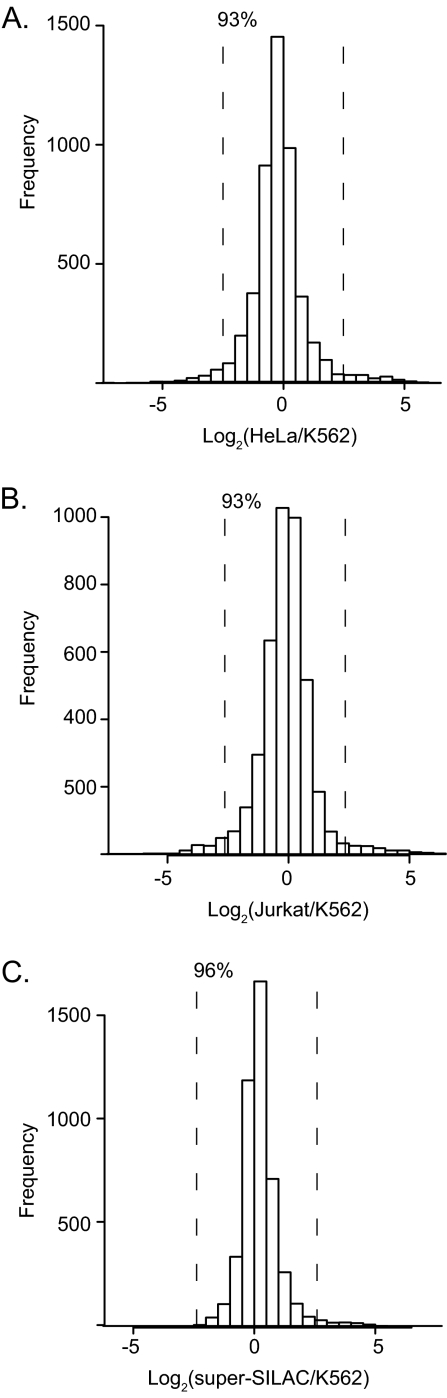
Deep proteomic analysis of mammalian cell lines would yield an inventory of the building blocks of the most commonly used systems in biological research. Mass spectrometry-based proteomics can identify and quantify proteins in a global and unbiased manner and can highlight the cellular processes that are altered between such systems. We analyzed 11 human cell lines using an LTQ-Orbitrap family mass spectrometer with a "high field" Orbitrap mass analyzer with improved resolution and sequencing speed. We identified a total of 11,731 proteins, and on average 10,361 ± 120 proteins in each cell line. This very high proteome coverage enabled analysis of a broad range of processes and functions. Despite the distinct origins of the cell lines, our quantitative results showed surprisingly high similarity in terms of expressed proteins. Nevertheless, this global similarity of the proteomes did not imply equal expression levels of individual proteins across the 11 cell lines, as we found significant differences in expression levels for an estimated two-third of them. The variability in cellular expression levels was similar for low and high abundance proteins, and even many of the most highly expressed proteins with household roles showed significant differences between cells. Metabolic pathways, which have high redundancy, exhibited variable expression, whereas basic cellular functions such as the basal transcription machinery varied much less. We harness knowledge of these cell line proteomes for the construction of a broad coverage "super-SILAC" quantification standard. Together with the accompanying paper (Schaab, C. MCP 2012, PMID: 22301388) (17) these data can be used to obtain reference expression profiles for proteins of interest both within and across cell line proteomes.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
