

Defective retinal vascular endothelial cell development as a consequence of impaired integrin αVβ8-mediated activation of transforming growth factor-β
- PMID: 22279205
- PMCID: PMC3578416
- DOI: 10.1523/JNEUROSCI.5648-11.2012
Defective retinal vascular endothelial cell development as a consequence of impaired integrin αVβ8-mediated activation of transforming growth factor-β
Erratum in
- J Neurosci. 2012 Apr 11;32(15):5351
Abstract
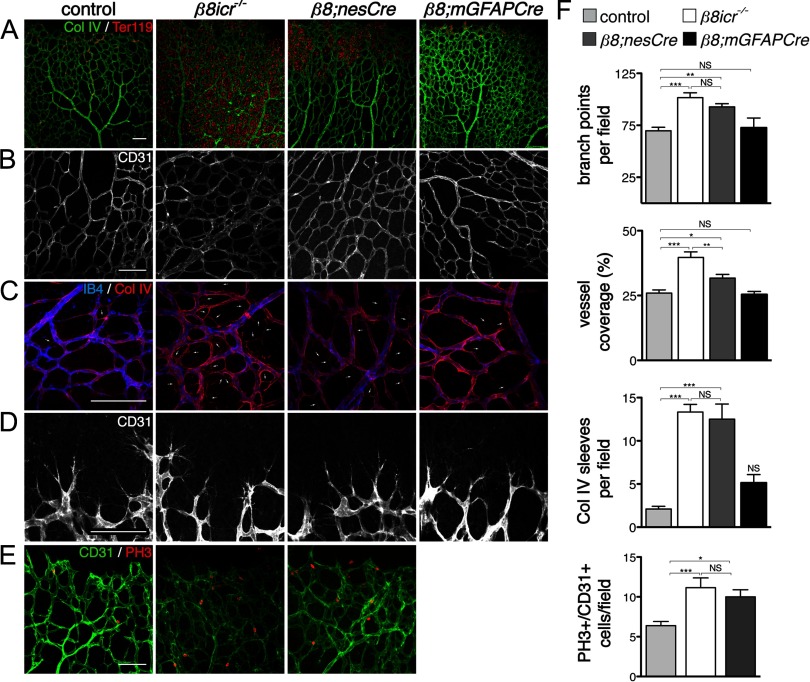
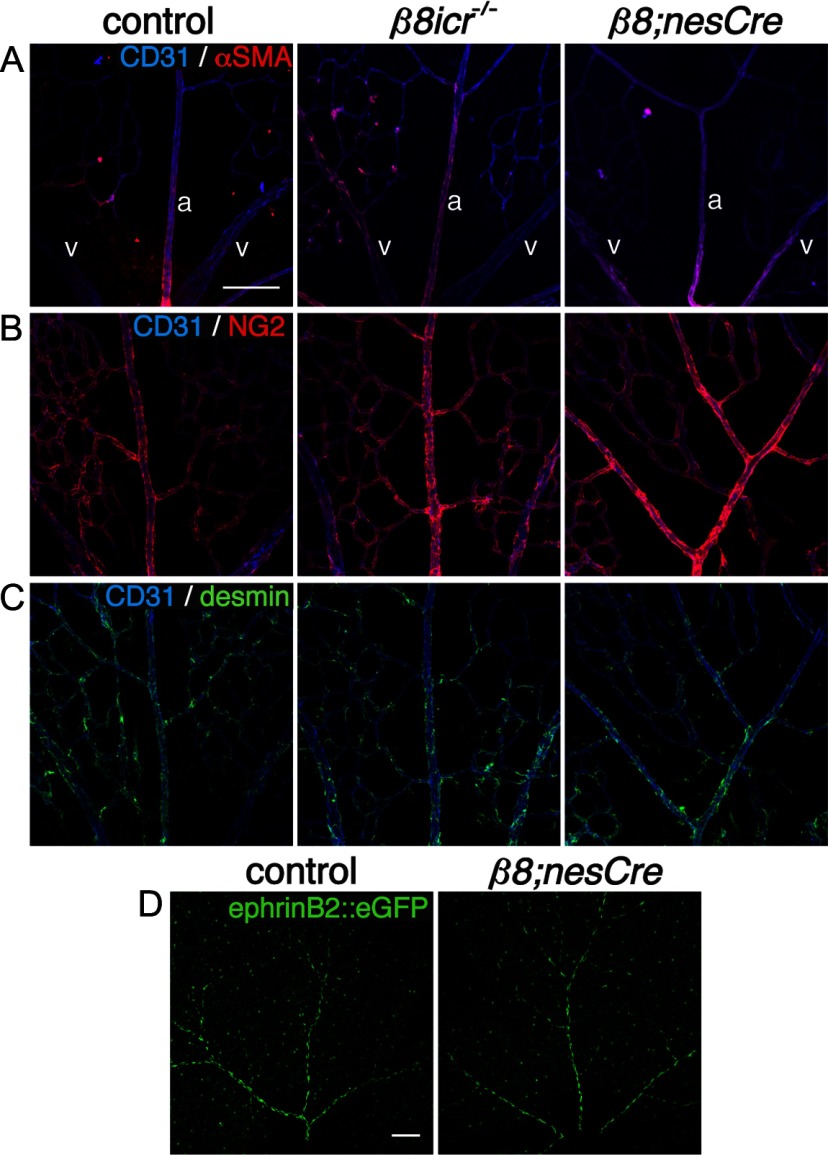
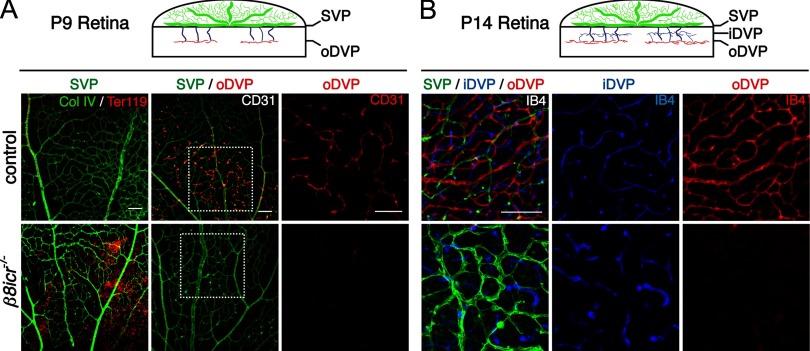
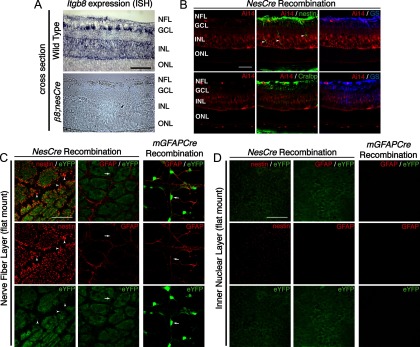
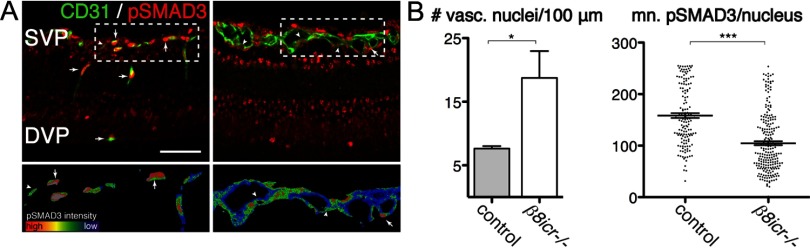
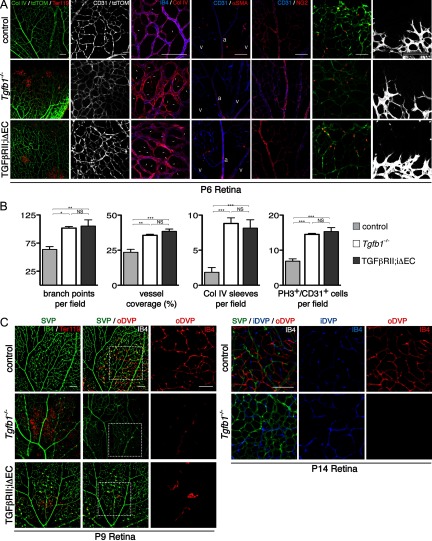
Deletions of the genes encoding the integrin αVβ8 (Itgav, Itgb8) have been shown to result in abnormal vascular development in the CNS, including prenatal and perinatal hemorrhage. Other work has indicated that a major function of this integrin in vivo is to promote TGFβ activation. In this paper, we show that Itgb8 mRNA is strongly expressed in murine Müller glia and retinal ganglion cells, but not astrocytes. We further show that Itgb8 deletion in the entire retina severely perturbs development of the murine retinal vasculature, elevating vascular branch point density and vascular coverage in the superficial vascular plexus, while severely impairing formation of the deep vascular plexus. The stability of the mutant vasculature is also impaired as assessed by the presence of hemorrhage and vascular basal lamina sleeves lacking endothelial cells. Specific deletion of Itgb8 in Müller glia and neurons, but not deletion in astrocytes, recapitulates the phenotype observed following Itgb8 in the entire retina. Consistent with αVβ8's role in TGFβ1 activation, we show that retinal deletion of Tgfb1 results in very similar retinal vascular abnormalities. The vascular deficits appear to reflect impaired TGFβ signaling in vascular endothelial cells because retinal deletion of Itgb8 reduces phospho-SMAD3 in endothelial cells and endothelial cell-specific deletion of the TGFβRII gene recapitulates the major deficits observed in the Itgb8 and TGFβ1 mutants. Of special interest, the retinal vascular phenotypes observed in each mutant are remarkably similar to those of others following inhibition of neuropilin-1, a receptor previously implicated in TGFβ activation and signaling.
Figures
References
-
- Adams RH, Klein R. Eph receptors and ephrin ligands. essential mediators of vascular development. Trends Cardiovasc Med. 2000;10:183–188. - PubMed
-
- Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. - PubMed
-
- Beránek M, Kanková K, Benes P, Izakovicová-Hollá L, Znojil V, Hájek D, Vlková E, Vácha J. Polymorphism R25P in the gene encoding transforming growth factor-beta (TGF-beta1) is a newly identified risk factor for proliferative diabetic retinopathy. Am J Med Genet. 2002;109:278–283. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous