

Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism
- PMID: 22284186
- PMCID: PMC3269786
- DOI: 10.1016/j.neuron.2011.10.038
Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism
Abstract
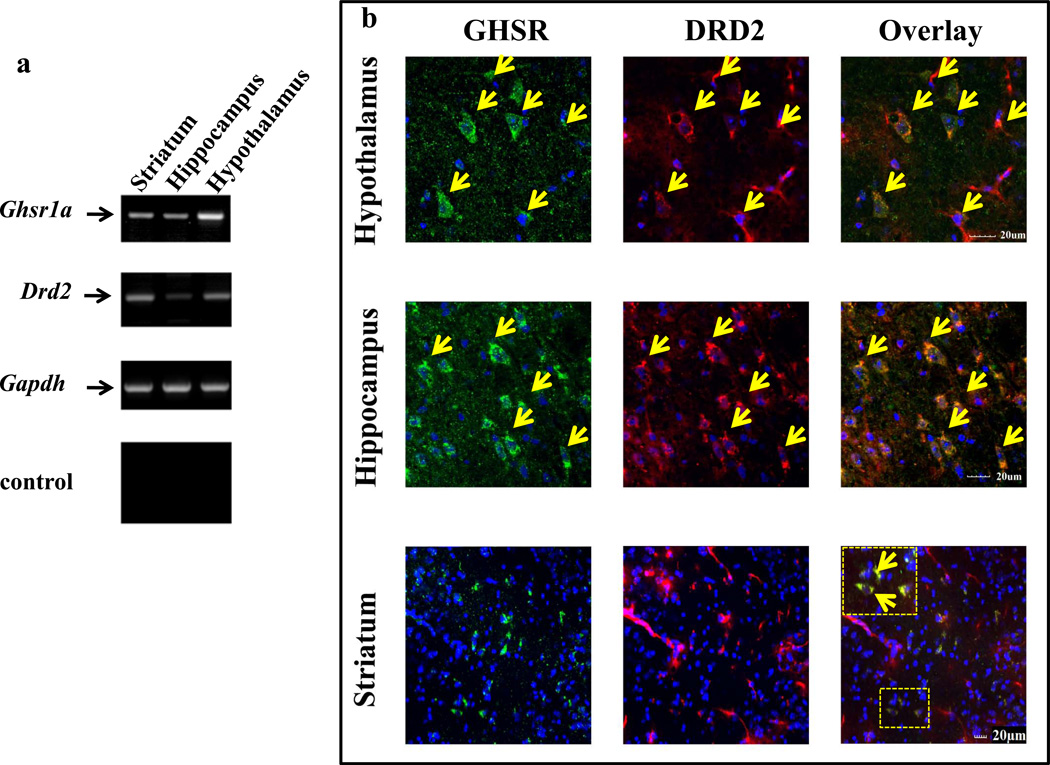
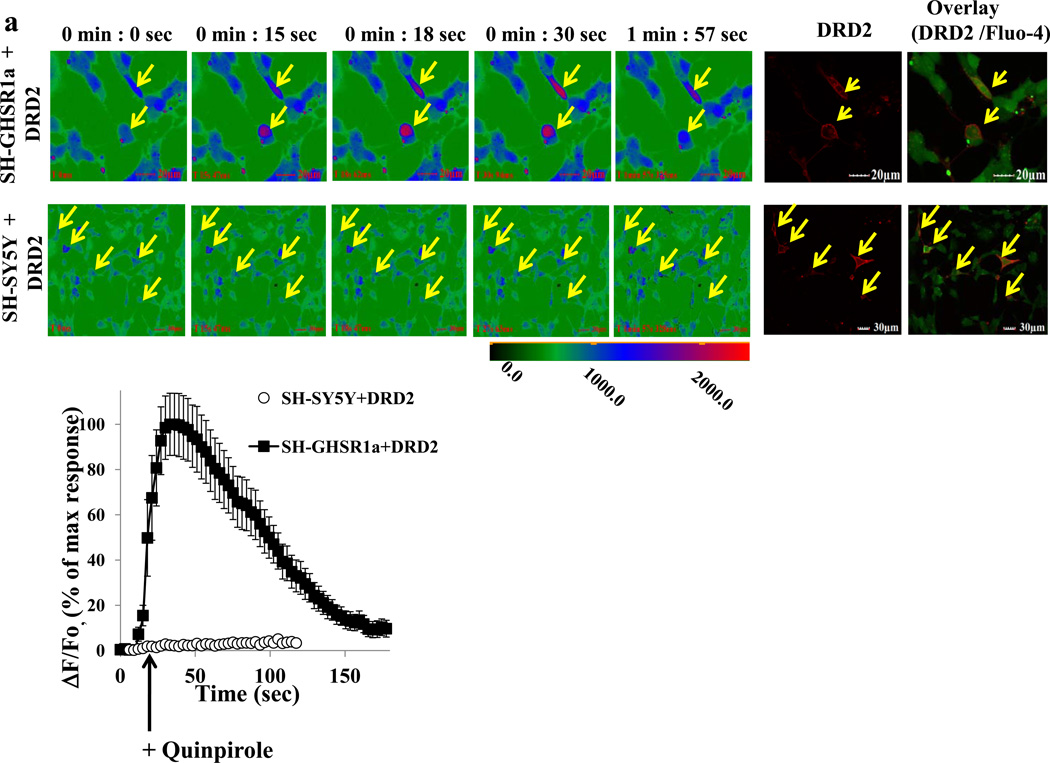
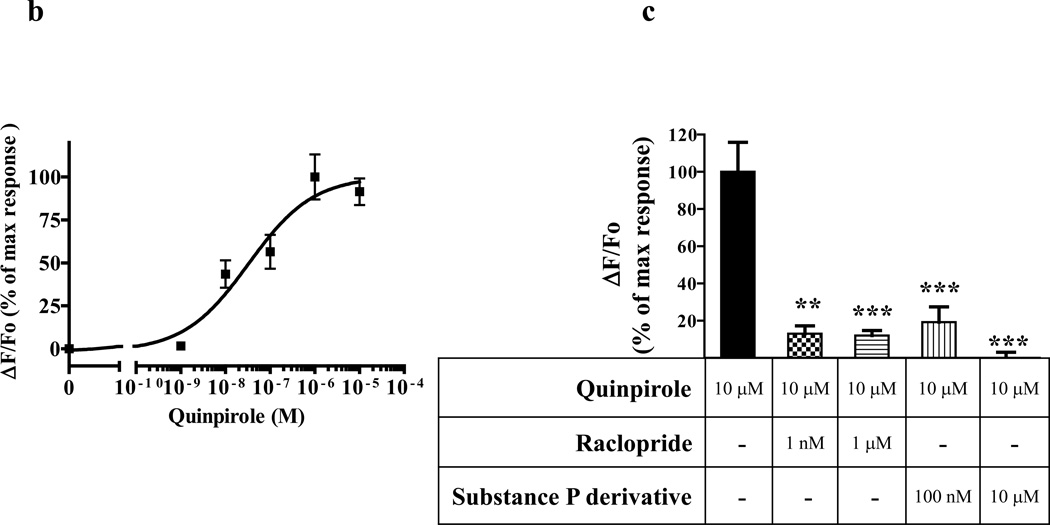
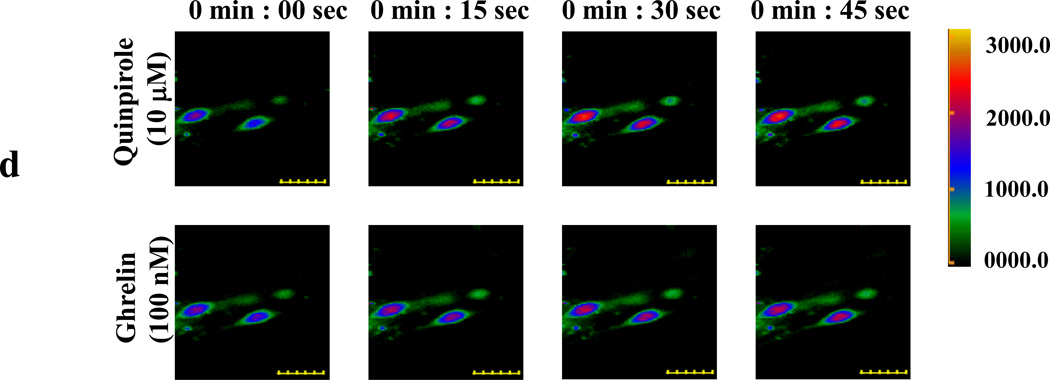
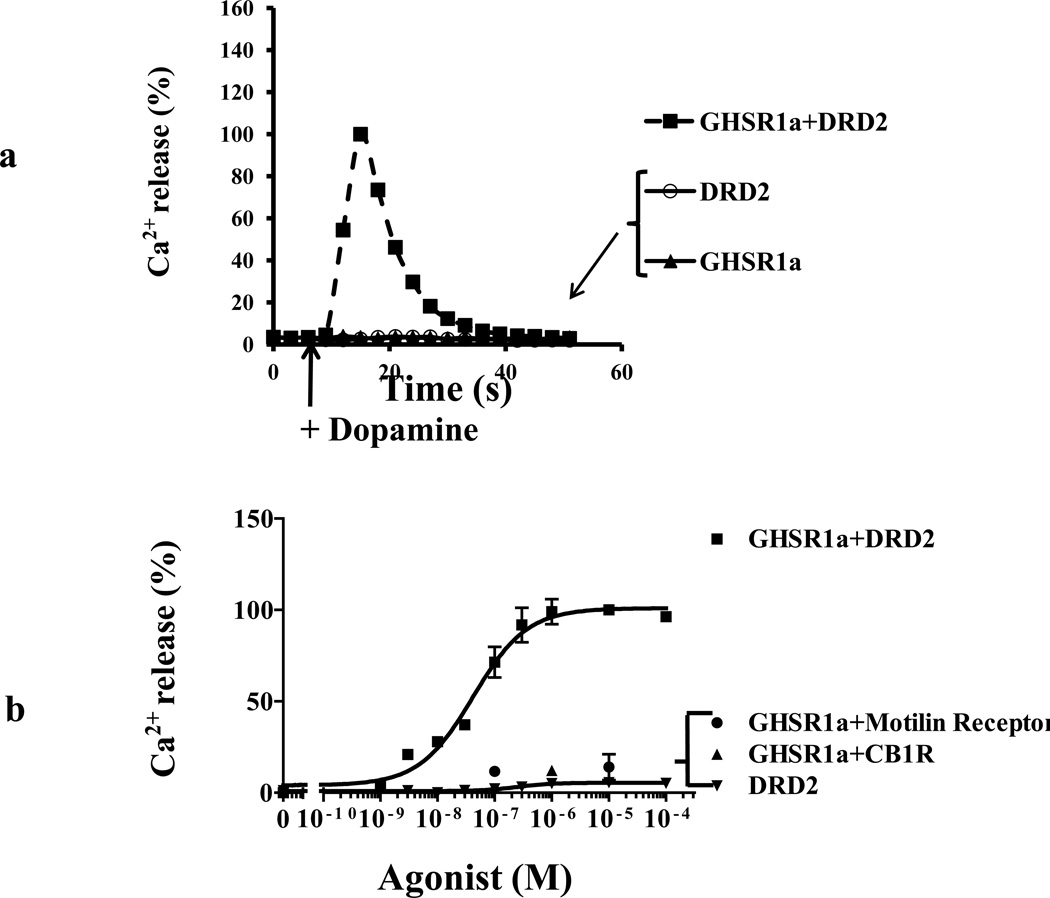
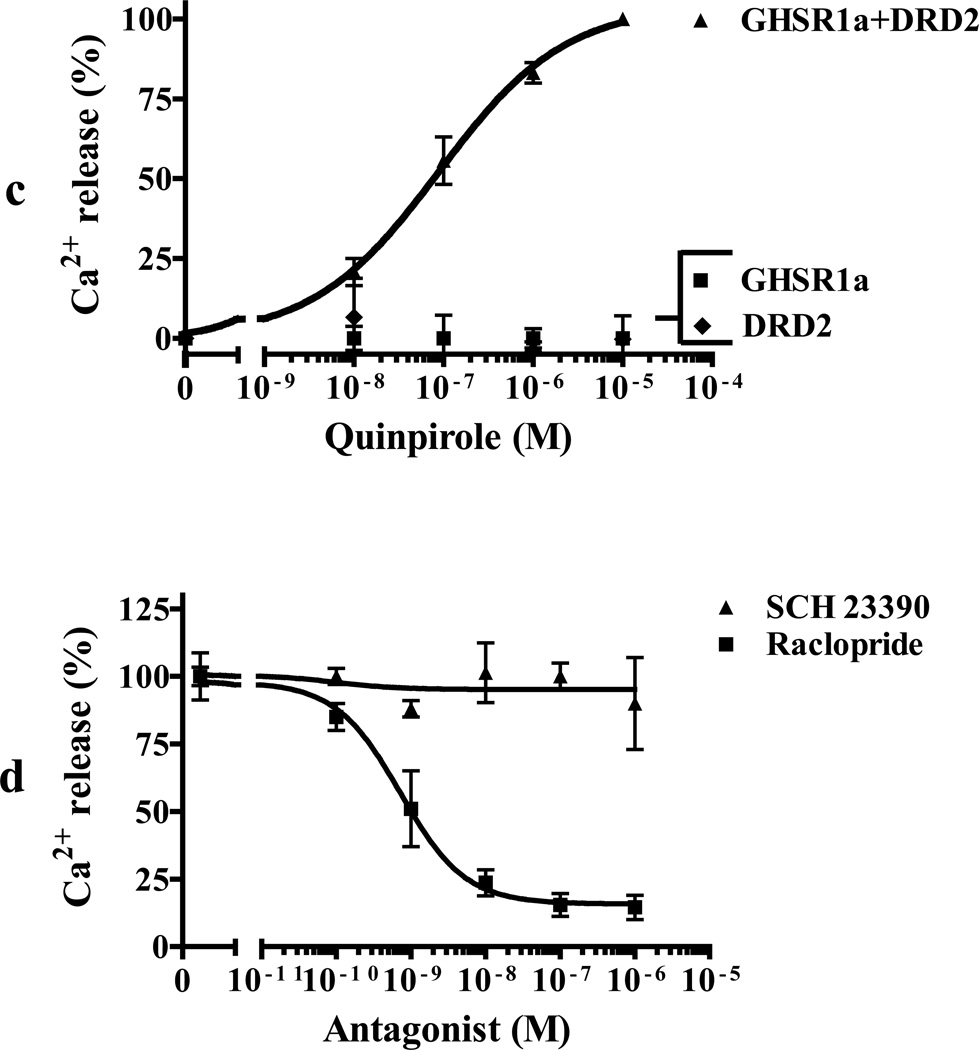
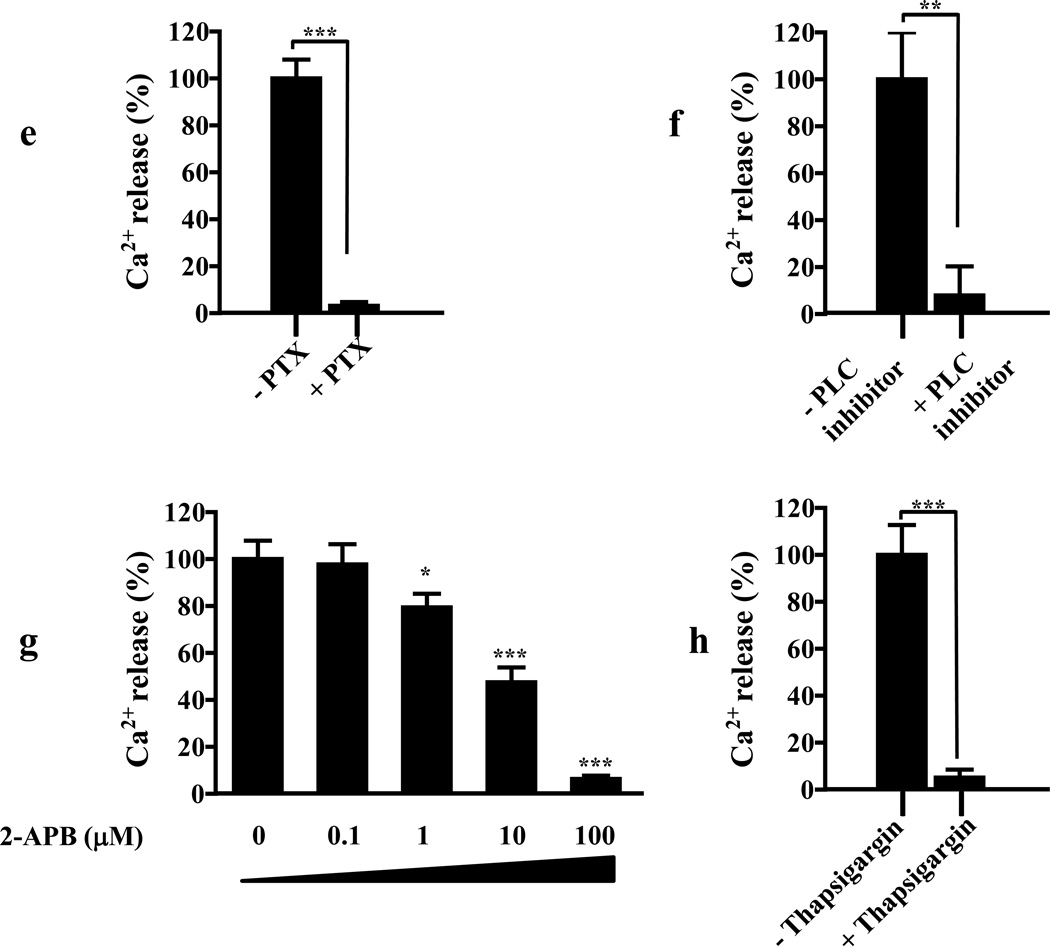
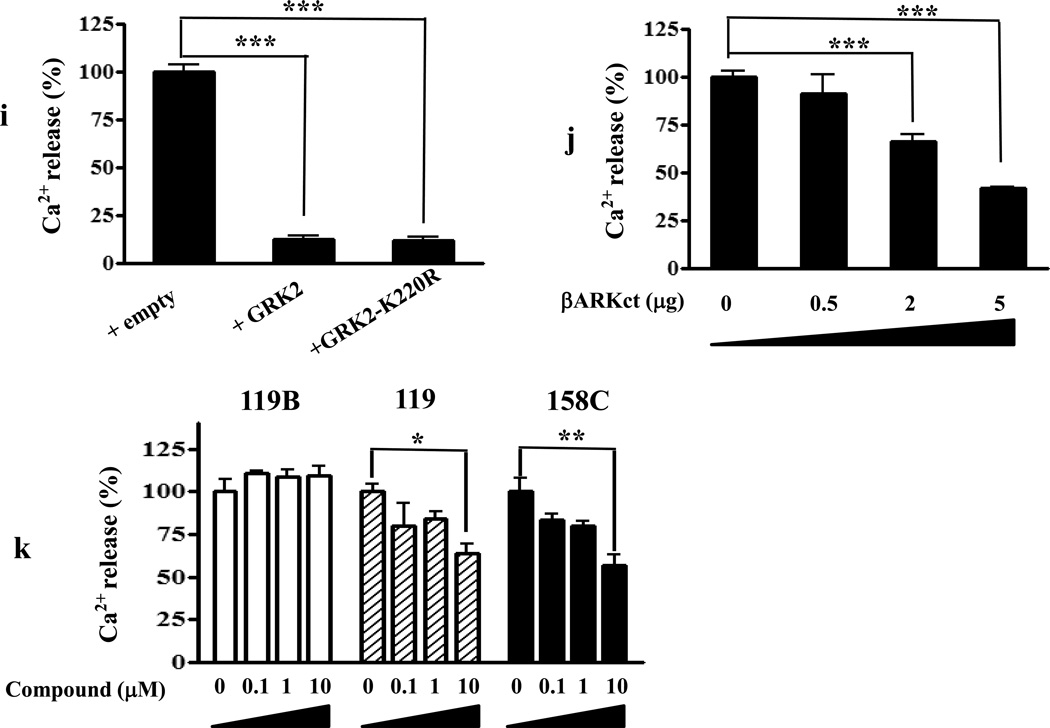
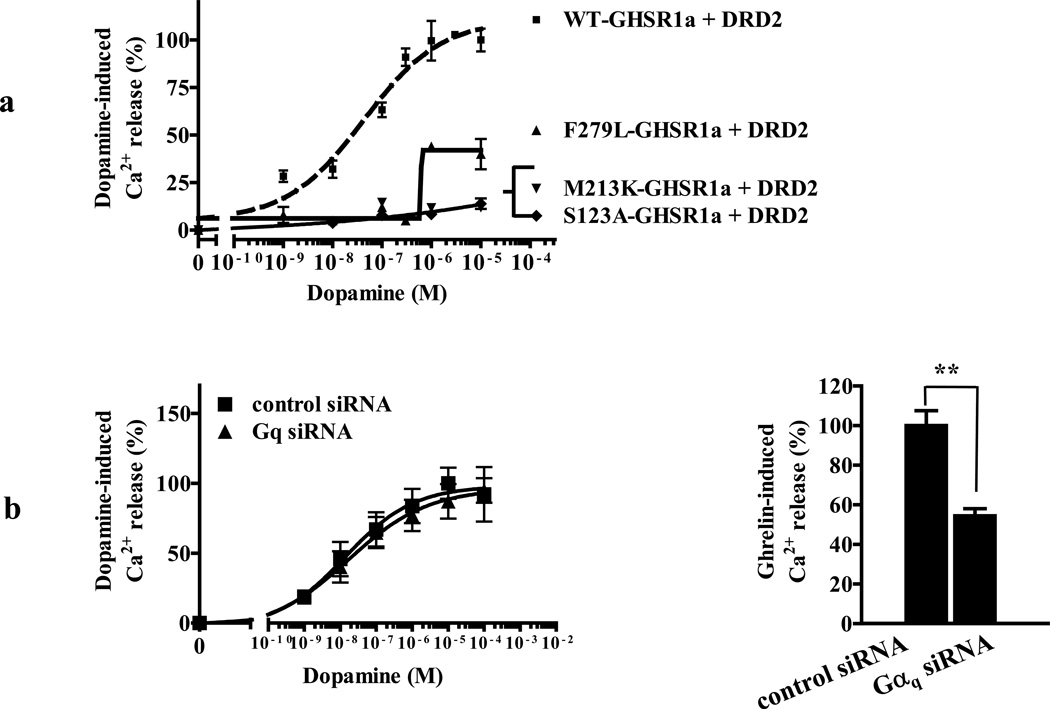
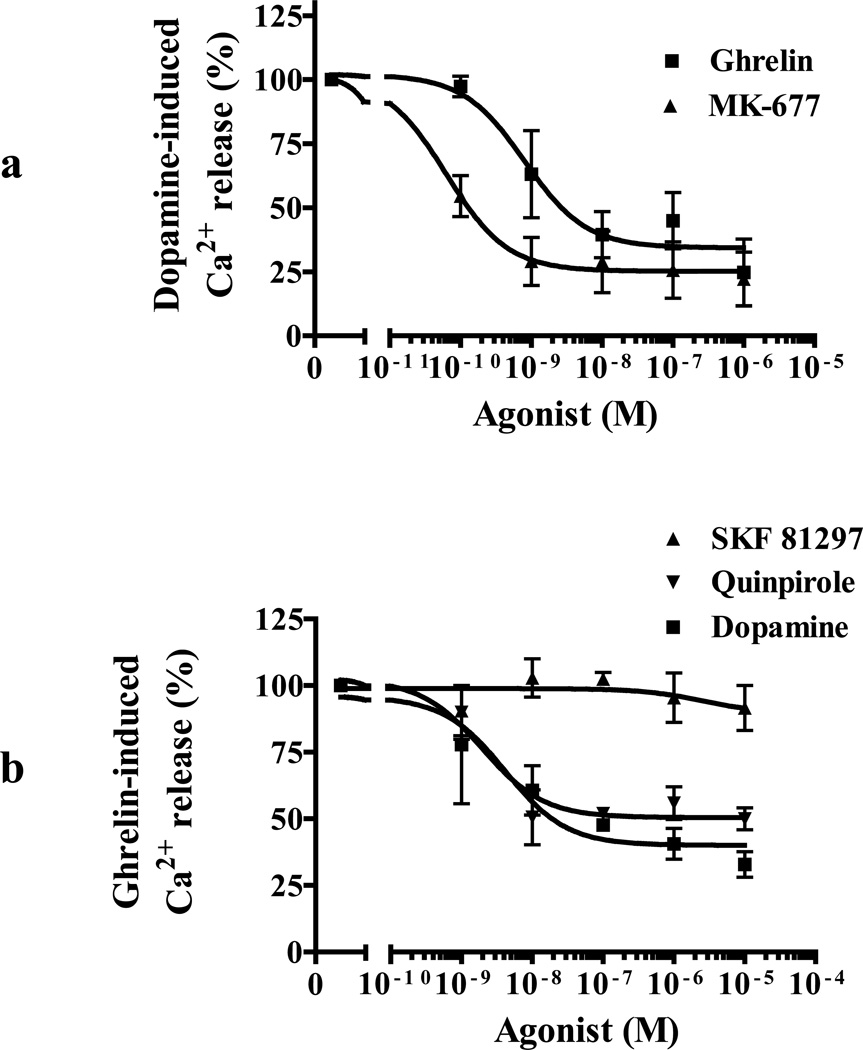
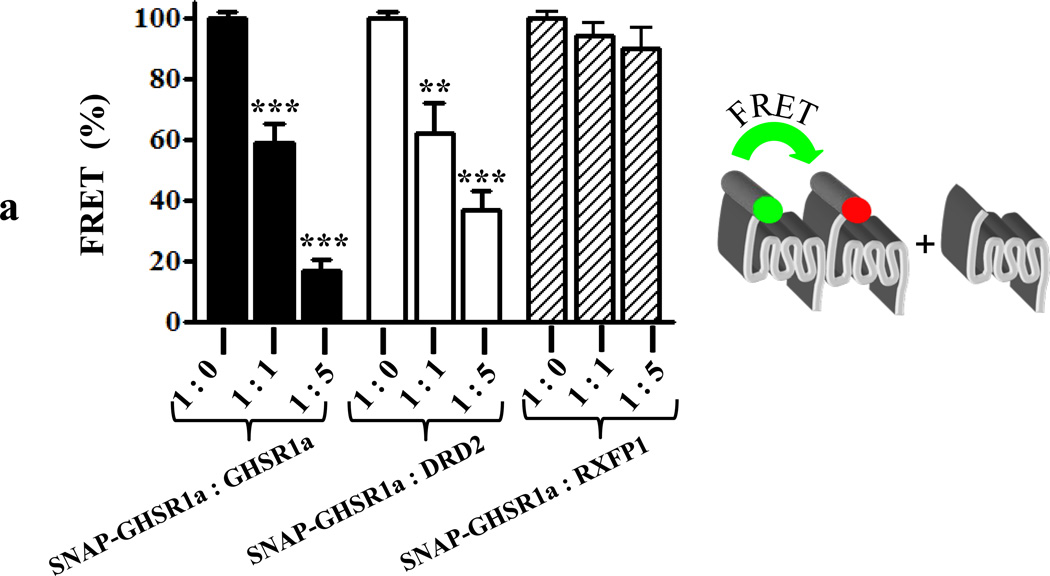
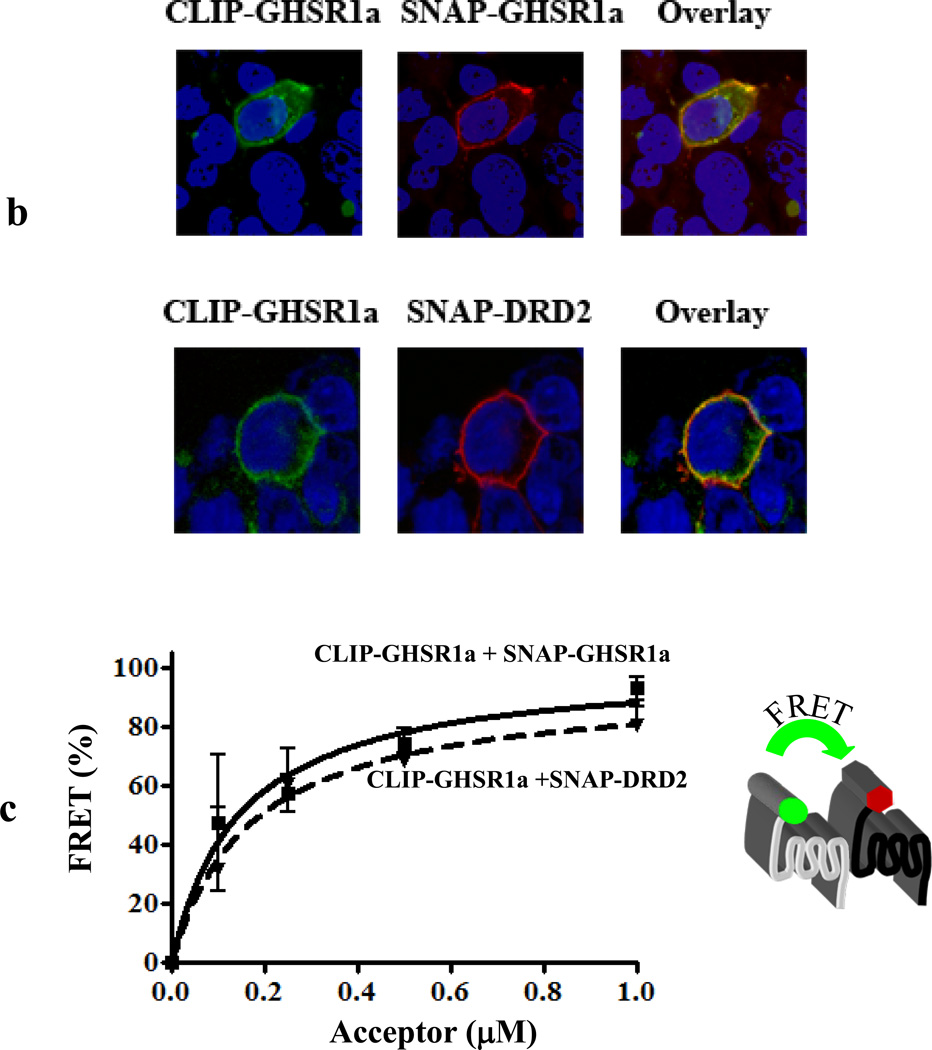
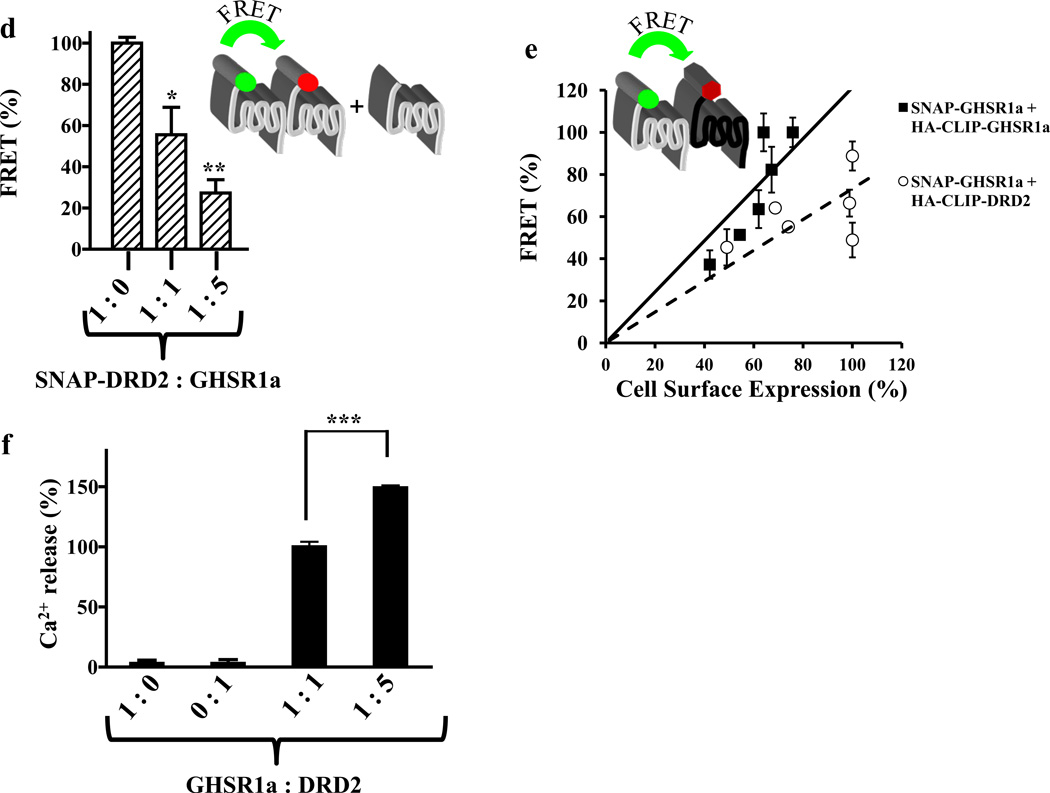
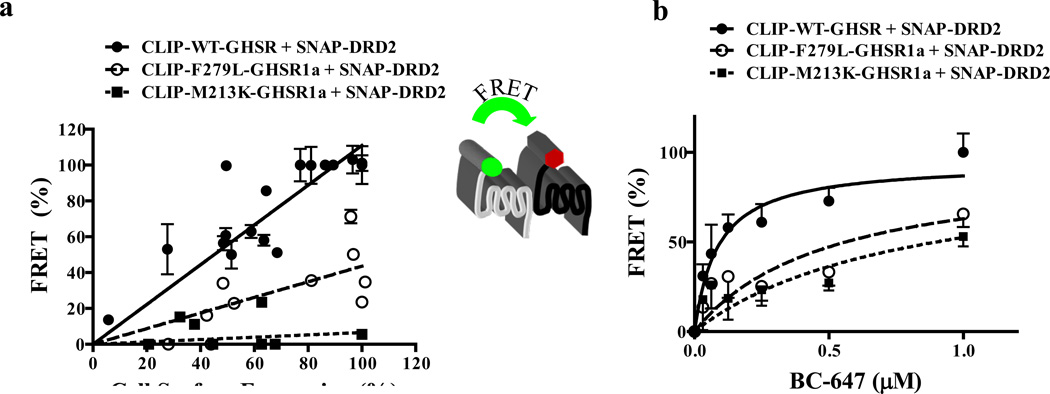
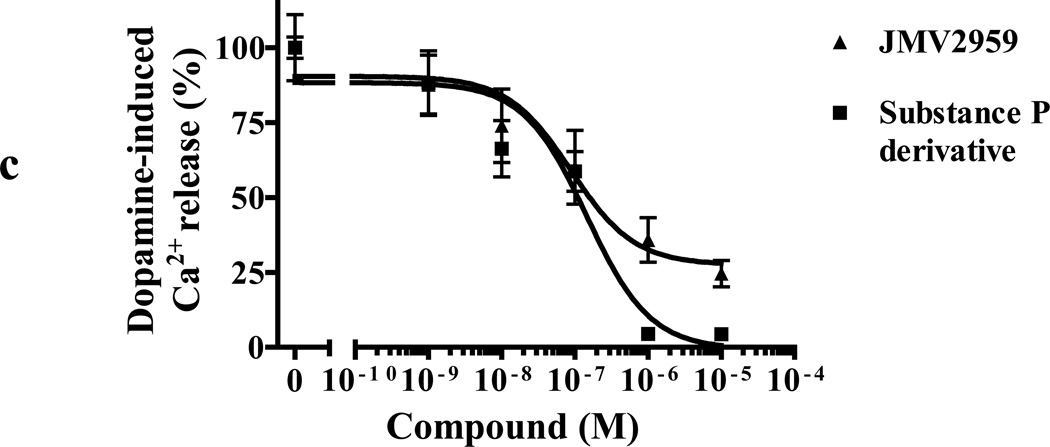
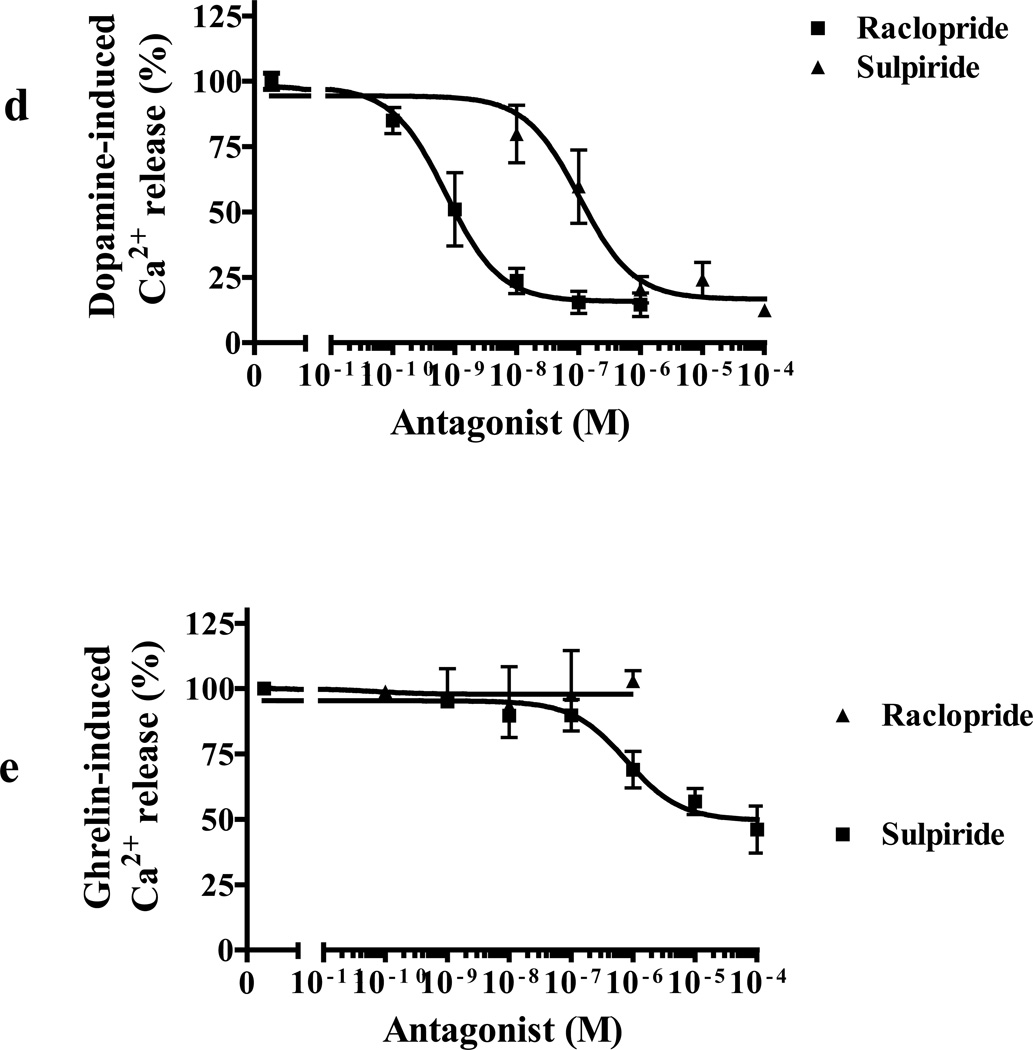
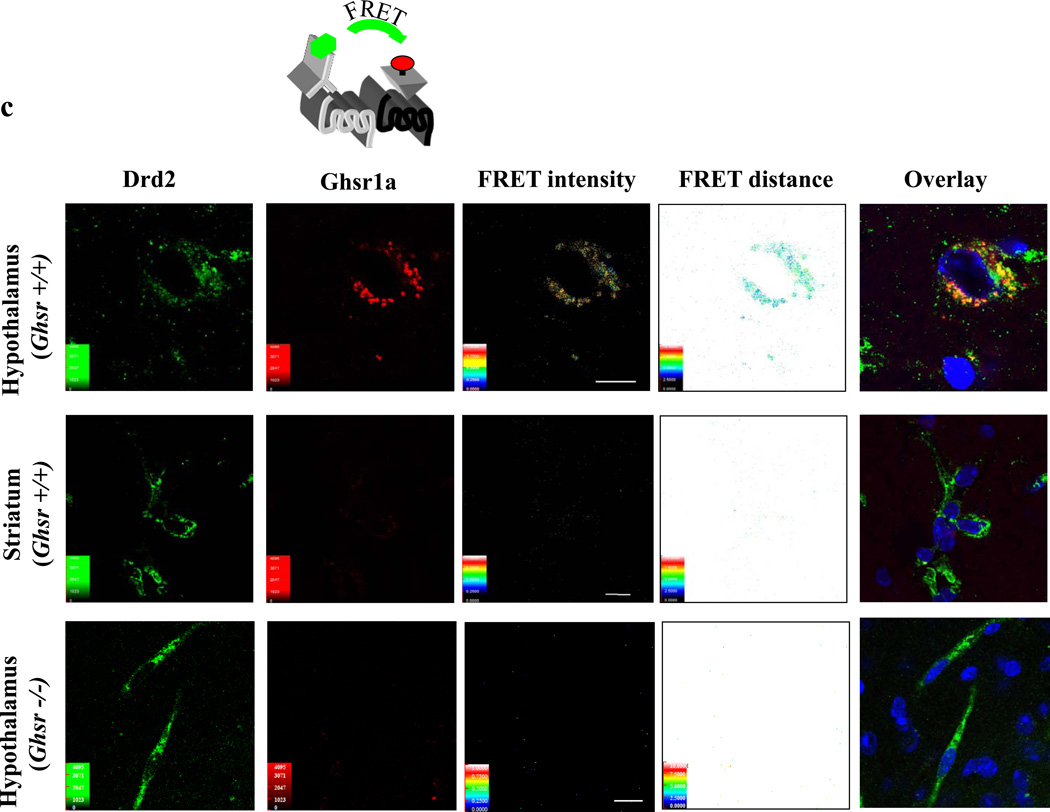
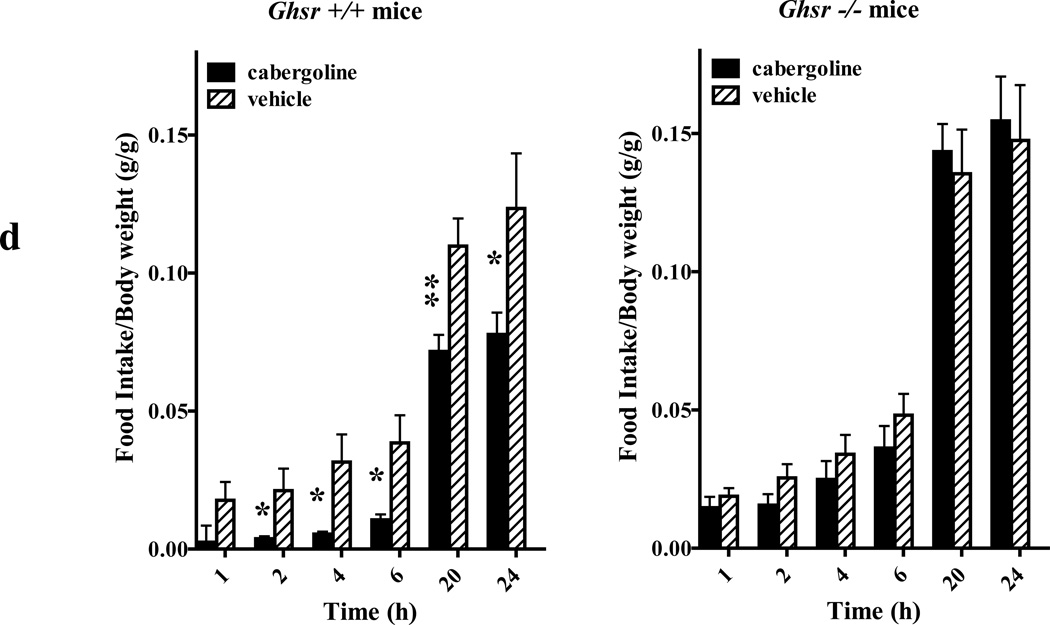
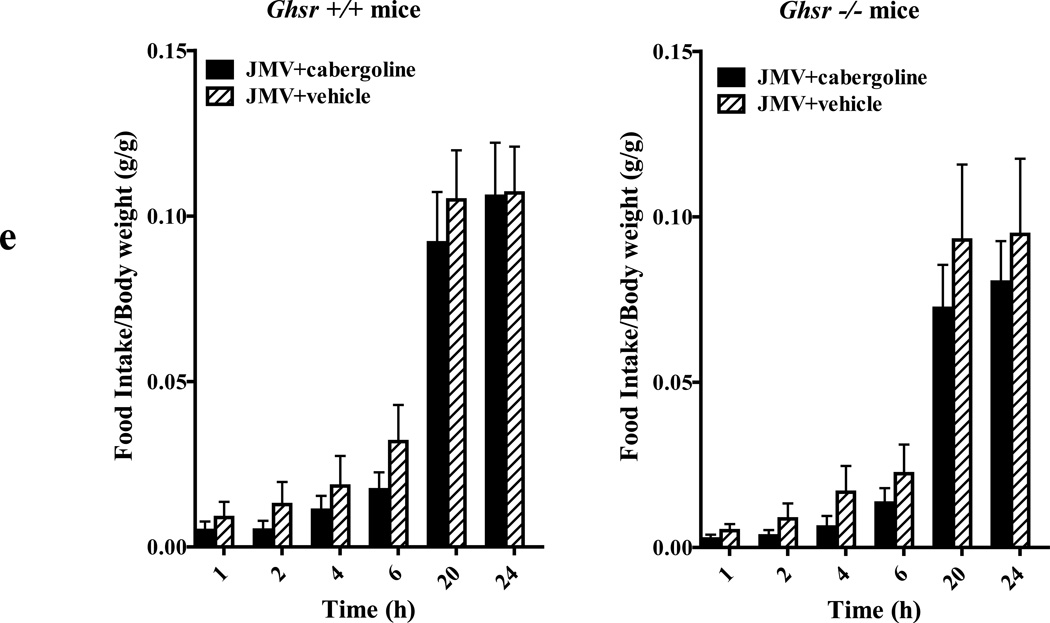
We identified subsets of neurons in the brain that coexpress the dopamine receptor subtype-2 (DRD2) and the ghrelin receptor (GHSR1a). Combination of FRET confocal microscopy and Tr-FRET established the presence of GHSR1a:DRD2 heteromers in hypothalamic neurons. To interrogate function, mice were treated with the selective DRD2 agonist cabergoline, which produced anorexia in wild-type and ghrelin⁻/⁻ mice; intriguingly, ghsr⁻/⁻ mice were refractory illustrating dependence on GHSR1a, but not ghrelin. Elucidation of mechanism showed that formation of GHSR1a:DRD2 heteromers allosterically modifies canonical DRD2 dopamine signaling resulting in Gβγ subunit-dependent mobilization of [Ca²⁺](i) independent of GHSR1a basal activity. By targeting the interaction between GHSR1a and DRD2 in wild-type mice with a highly selective GHSR1a antagonist (JMV2959) cabergoline-induced anorexia was blocked. Inhibiting dopamine signaling in subsets of neurons with a GHSR1a antagonist has profound therapeutic implications by providing enhanced selectivity because neurons expressing DRD2 alone would be unaffected.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures


Comment in
-
Food for thought: the physiological relevance of ghrelin and dopamine D2 receptor heterodimerization in the regulation of appetite.Neuron. 2012 Jan 26;73(2):210-1. doi: 10.1016/j.neuron.2012.01.004. Neuron. 2012. PMID: 22284175
References
-
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of Gbetagamma-subunit signaling with small molecules. Science. 2006;312:443–446. - PubMed
-
- Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. - PubMed
-
- Button D, Brownstein M. Aequorin-expressing mammalian cell lines used to report Ca2+ mobilization. Cell Calcium. 1993;14:663–671. - PubMed
-
- Canals M, Milligan G. Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed Mu opioid receptors. J Biol Chem. 2008;283:11424–11434. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
