

Sodium fluoride induces apoptosis in mouse embryonic stem cells through ROS-dependent and caspase- and JNK-mediated pathways
- PMID: 22285274
- PMCID: PMC3299821
- DOI: 10.1016/j.taap.2012.01.010
Sodium fluoride induces apoptosis in mouse embryonic stem cells through ROS-dependent and caspase- and JNK-mediated pathways
Abstract
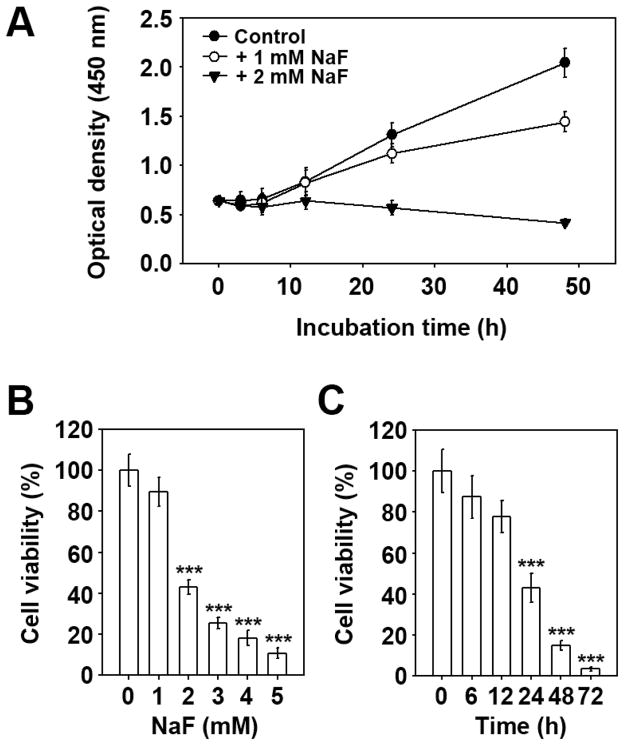
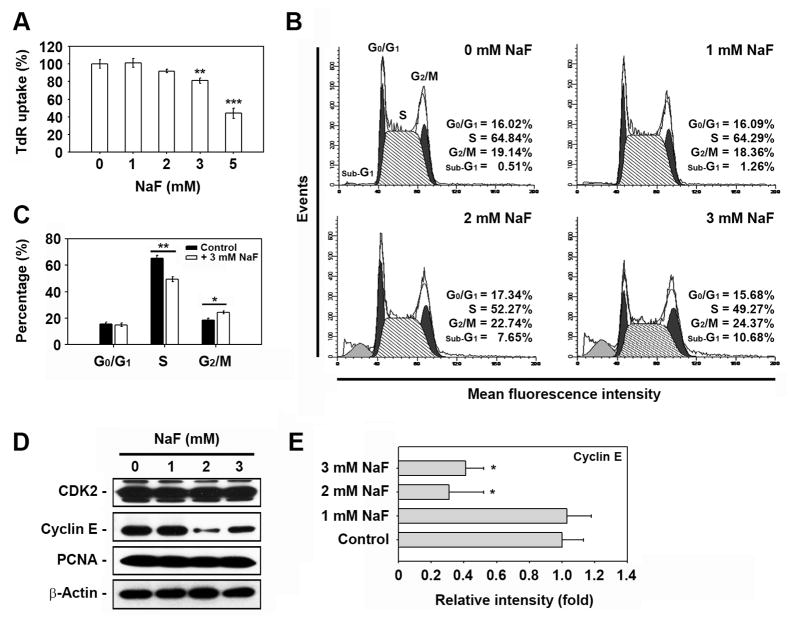
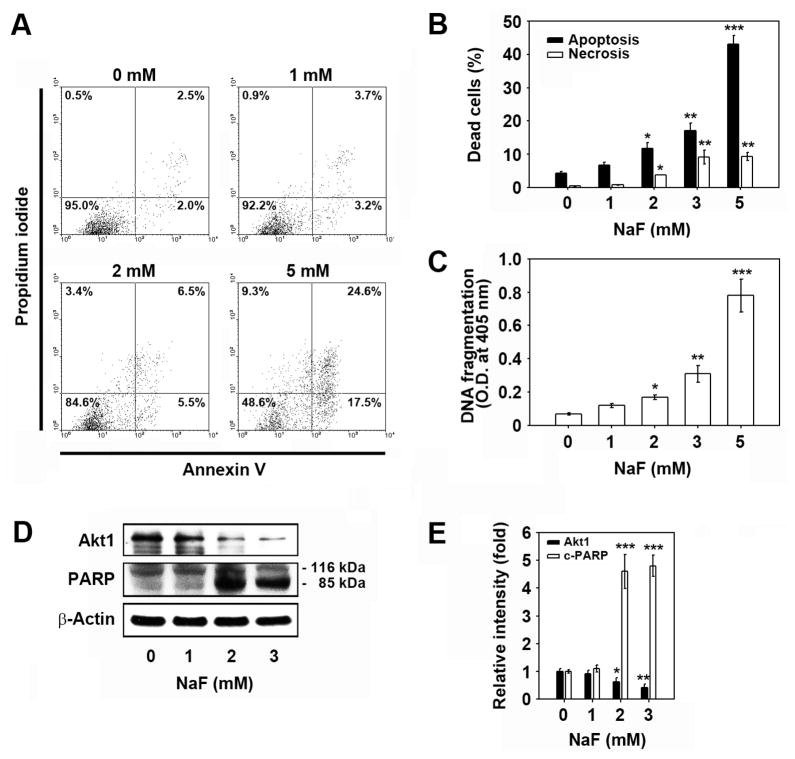
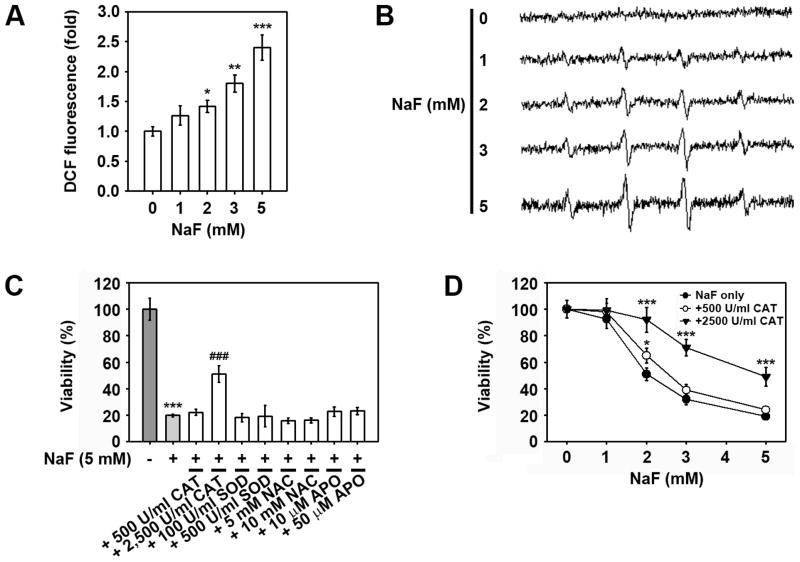
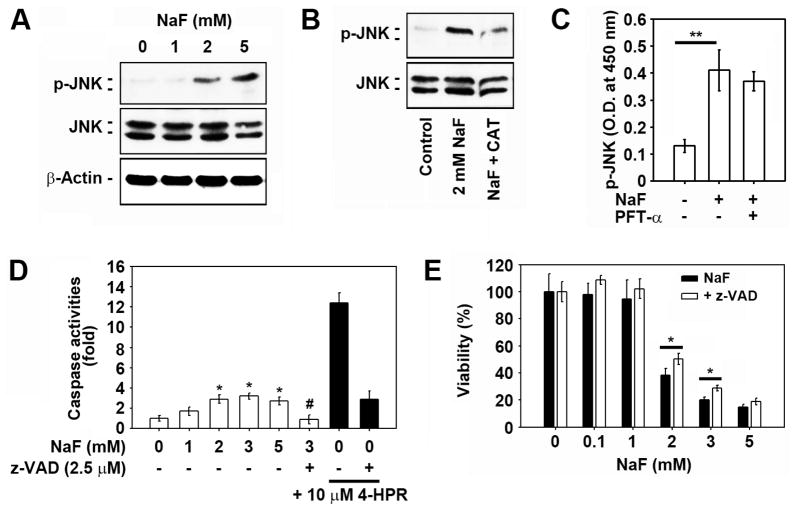
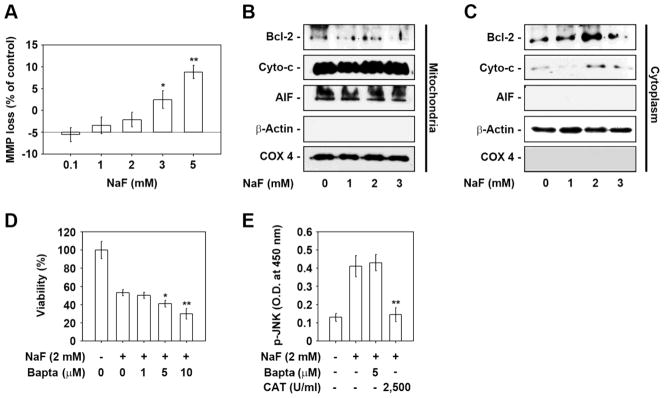
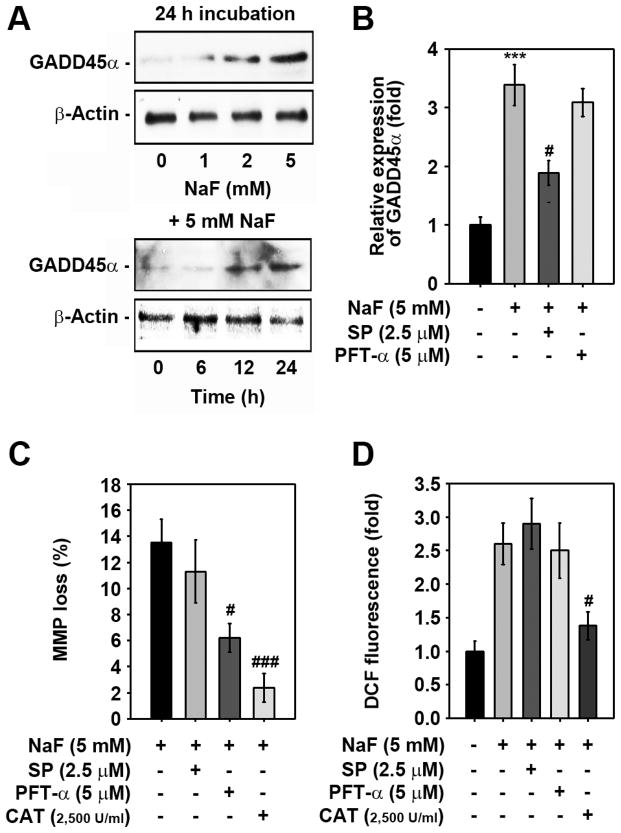
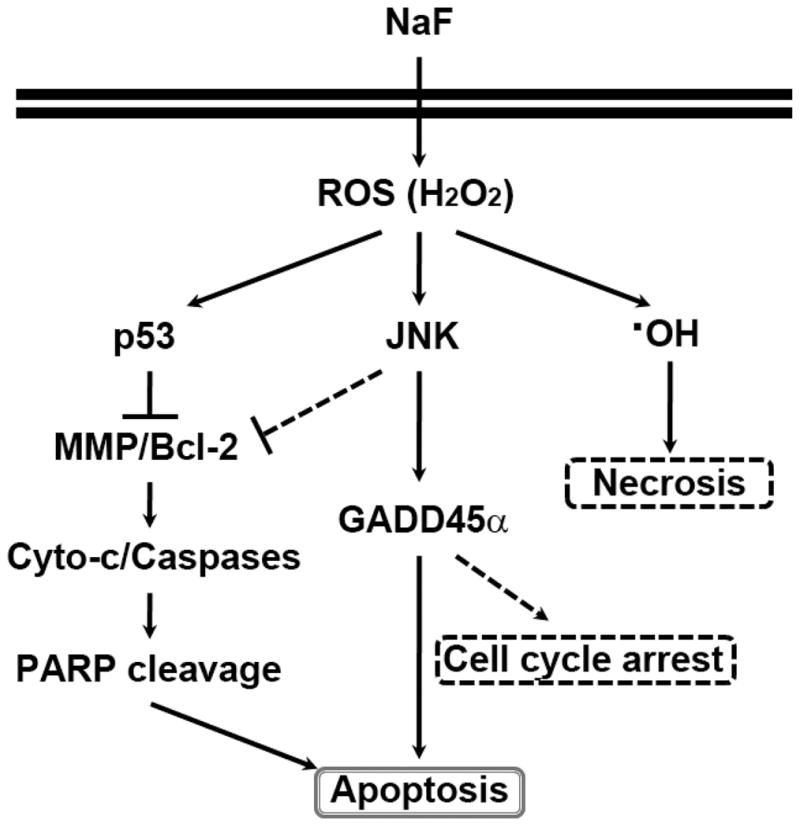
Sodium fluoride (NaF) is used as a source of fluoride ions in diverse applications. Fluoride salt is an effective prophylactic for dental caries and is an essential element required for bone health. However, fluoride is known to cause cytotoxicity in a concentration-dependent manner. Further, no information is available on the effects of NaF on mouse embryonic stem cells (mESCs). We investigated the mode of cell death induced by NaF and the mechanisms involved. NaF treatment greater than 1mM reduced viability and DNA synthesis in mESCs and induced cell cycle arrest in the G(2)/M phase. The addition of NaF induced cell death mainly by apoptosis rather than necrosis. Catalase (CAT) treatment significantly inhibited the NaF-mediated cell death and also suppressed the NaF-mediated increase in phospho-c-Jun N-terminal kinase (p-JNK) levels. Pre-treatment with SP600125 or z-VAD-fmk significantly attenuated the NaF-mediated reduction in cell viability. In contrast, intracellular free calcium chelator, but not of sodium or calcium ion channel blockers, facilitated NaF-induced toxicity in the cells. A JNK specific inhibitor (SP600125) prevented the NaF-induced increase in growth arrest and the DNA damage-inducible protein 45α. Further, NaF-mediated loss of mitochondrial membrane potential was apparently inhibited by pifithrin-α or CAT inhibitor. These findings suggest that NaF affects viability of mESCs in a concentration-dependent manner, where more than 1mM NaF causes apoptosis through hydroxyl radical-dependent and caspase- and JNK-mediated pathways.
Copyright © 2012 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures
References
-
- Barbier O, Arreola-Mendoza L, Del Razo LM. Molecular mechanisms of fluoride toxicity. Chem Biol Interact. 2010;188:319–333. - PubMed
-
- Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D. DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation. Free Radic Biol Med. 2002;33:691–702. - PubMed
-
- Ba Y, Zhu JY, Yang YJ, Yu B, Huang H, Wang C, Ren LJ, Cheng XM, Cui LX, Zhang YW. Serum calciotropic hormone levels, and dental fluorisis in children exposed to different concentrations of fluoride and iodine in dringking water. Chin Med J. 2010;123:675–679. - PubMed
-
- Bourgoin SG, Harbour D, Poubelle PE. Role of protein kinase C alpha, Arf, and cytoplasmic calcium transients in phospholipase D activation by sodium fluoride in osteoblast-like cells. J Bone Miner Res. 1996;11:1655–1665. - PubMed
-
- Buzalaf MA, Levy SM. Fluoride intake of children: considerations for dental caries and dental fluorosis. Monogr Oral Sci. 2011;22:1–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
