

Removing technical variability in RNA-seq data using conditional quantile normalization
- PMID: 22285995
- PMCID: PMC3297825
- DOI: 10.1093/biostatistics/kxr054
Removing technical variability in RNA-seq data using conditional quantile normalization
Abstract
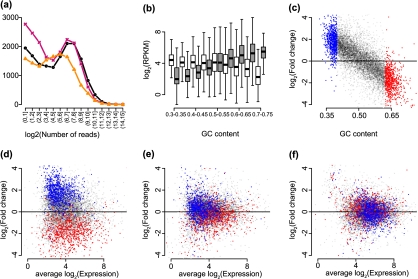
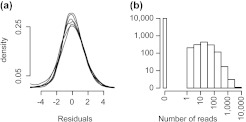
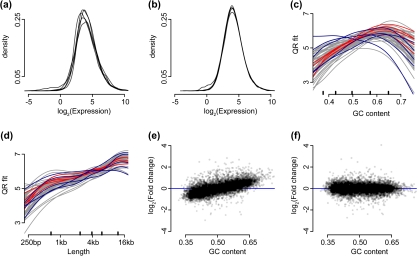
The ability to measure gene expression on a genome-wide scale is one of the most promising accomplishments in molecular biology. Microarrays, the technology that first permitted this, were riddled with problems due to unwanted sources of variability. Many of these problems are now mitigated, after a decade's worth of statistical methodology development. The recently developed RNA sequencing (RNA-seq) technology has generated much excitement in part due to claims of reduced variability in comparison to microarrays. However, we show that RNA-seq data demonstrate unwanted and obscuring variability similar to what was first observed in microarrays. In particular, we find guanine-cytosine content (GC-content) has a strong sample-specific effect on gene expression measurements that, if left uncorrected, leads to false positives in downstream results. We also report on commonly observed data distortions that demonstrate the need for data normalization. Here, we describe a statistical methodology that improves precision by 42% without loss of accuracy. Our resulting conditional quantile normalization algorithm combines robust generalized regression to remove systematic bias introduced by deterministic features such as GC-content and quantile normalization to correct for global distortions.
Figures

References
-
- Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
