

Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification
- PMID: 22288382
- PMCID: PMC3319072
- DOI: 10.1021/pr2008175
Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification
Abstract
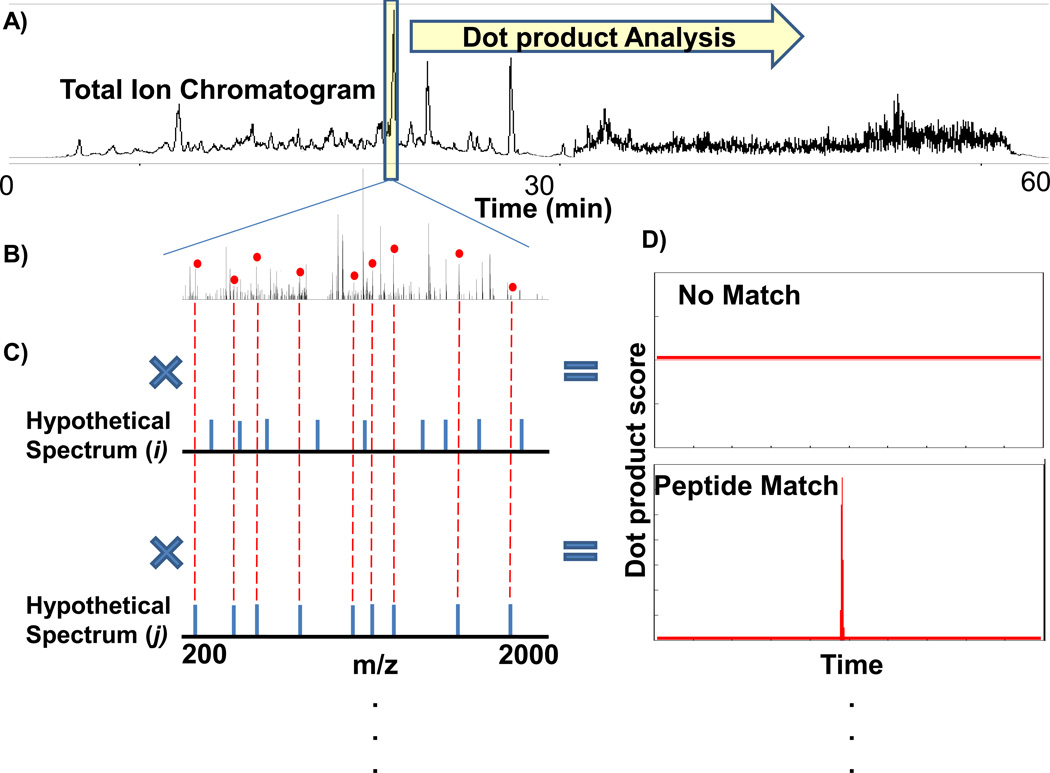
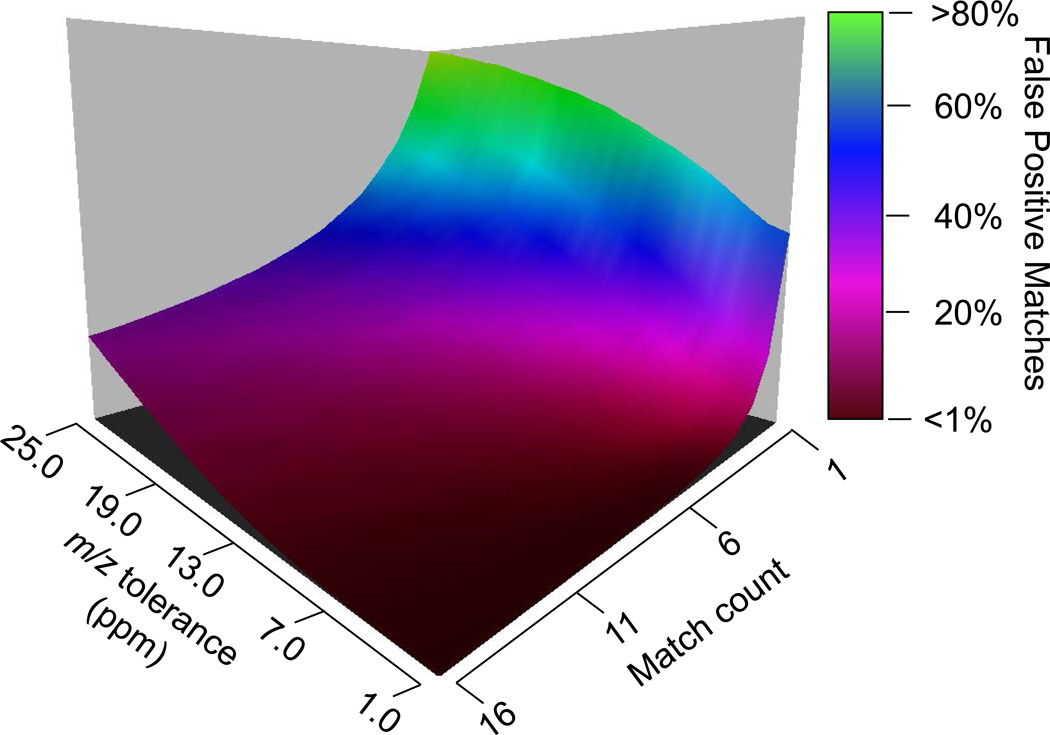
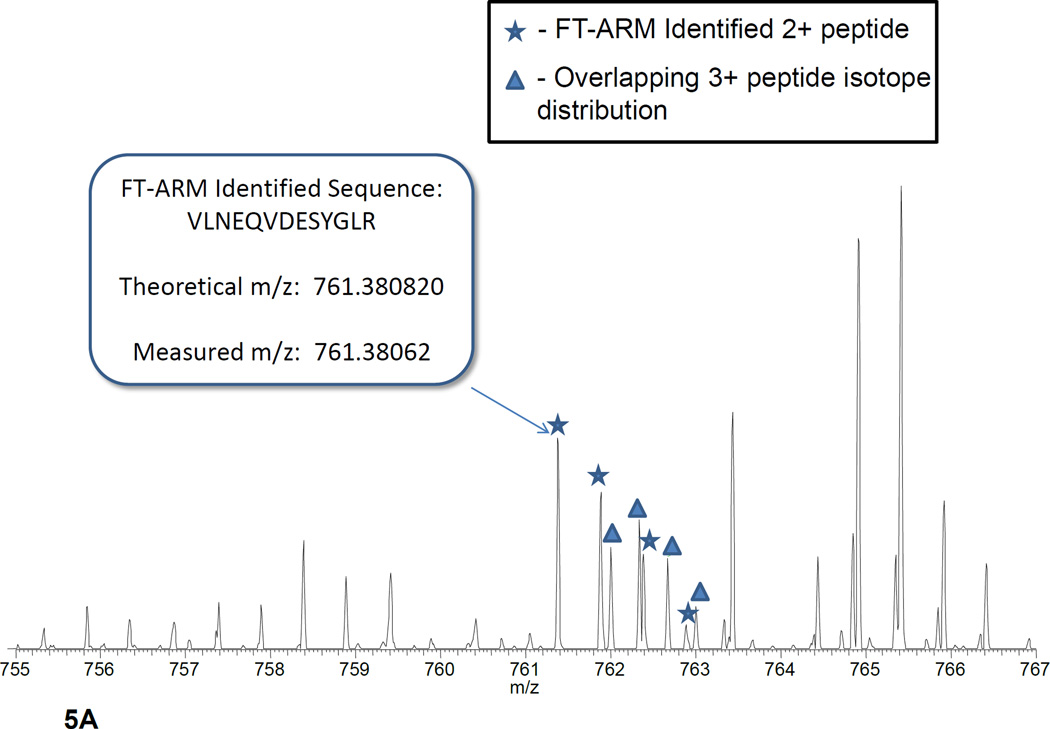
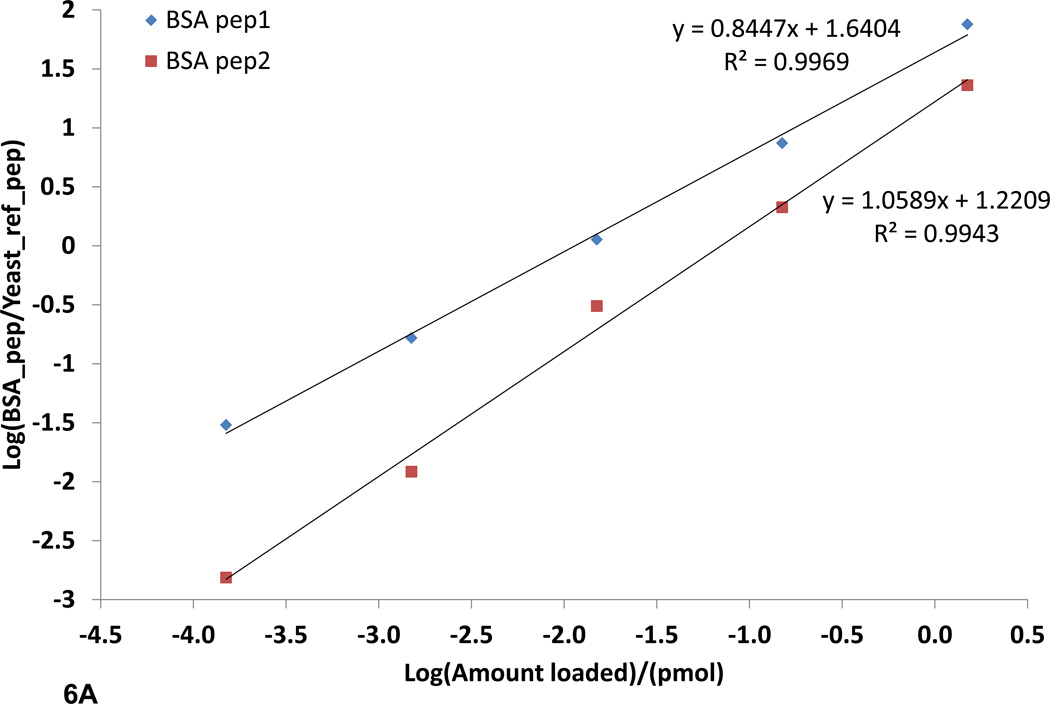
Fourier transform-all reaction monitoring (FT-ARM) is a novel approach for the identification and quantification of peptides that relies upon the selectivity of high mass accuracy data and the specificity of peptide fragmentation patterns. An FT-ARM experiment involves continuous, data-independent, high mass accuracy MS/MS acquisition spanning a defined m/z range. Custom software was developed to search peptides against the multiplexed fragmentation spectra by comparing theoretical or empirical fragment ions against every fragmentation spectrum across the entire acquisition. A dot product score is calculated against each spectrum to generate a score chromatogram used for both identification and quantification. Chromatographic elution profile characteristics are not used to cluster precursor peptide signals to their respective fragment ions. FT-ARM identifications are demonstrated to be complementary to conventional data-dependent shotgun analysis, especially in cases where the data-dependent method fails because of fragmenting multiple overlapping precursors. The sensitivity, robustness, and specificity of FT-ARM quantification are shown to be analogous to selected reaction monitoring-based peptide quantification with the added benefit of minimal assay development. Thus, FT-ARM is demonstrated to be a novel and complementary data acquisition, identification, and quantification method for the large scale analysis of peptides.
Figures





Similar articles
-
Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis.Mol Cell Proteomics. 2012 Jun;11(6):O111.016717. doi: 10.1074/mcp.O111.016717. Epub 2012 Jan 18. Mol Cell Proteomics. 2012. PMID: 22261725 Free PMC article.
-
Pepitome: evaluating improved spectral library search for identification complementarity and quality assessment.J Proteome Res. 2012 Mar 2;11(3):1686-95. doi: 10.1021/pr200874e. Epub 2012 Jan 27. J Proteome Res. 2012. PMID: 22217208 Free PMC article.
-
Multiplexed and data-independent tandem mass spectrometry for global proteome profiling.Mass Spectrom Rev. 2014 Nov-Dec;33(6):452-70. doi: 10.1002/mas.21400. Epub 2013 Nov 26. Mass Spectrom Rev. 2014. PMID: 24281846 Review.
-
Peptides quantification by liquid chromatography with matrix-assisted laser desorption/ionization and selected reaction monitoring detection.J Proteome Res. 2012 Oct 5;11(10):4972-82. doi: 10.1021/pr300514u. Epub 2012 Aug 31. J Proteome Res. 2012. PMID: 22897511
-
Peptide identification by tandem mass spectrometry with alternate fragmentation modes.Mol Cell Proteomics. 2012 Sep;11(9):550-7. doi: 10.1074/mcp.R112.018556. Epub 2012 May 17. Mol Cell Proteomics. 2012. PMID: 22595789 Free PMC article. Review.
Cited by
-
Multiplexed peptide analysis using data-independent acquisition and Skyline.Nat Protoc. 2015 Jun;10(6):887-903. doi: 10.1038/nprot.2015.055. Epub 2015 May 21. Nat Protoc. 2015. PMID: 25996789 Free PMC article.
-
Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries.Mol Cell Proteomics. 2020 Jul;19(7):1088-1103. doi: 10.1074/mcp.P119.001913. Epub 2020 Apr 20. Mol Cell Proteomics. 2020. PMID: 32312845 Free PMC article.
-
Comparison of data acquisition strategies on quadrupole ion trap instrumentation for shotgun proteomics.J Am Soc Mass Spectrom. 2014 Dec;25(12):2048-59. doi: 10.1007/s13361-014-0981-1. Epub 2014 Sep 27. J Am Soc Mass Spectrom. 2014. PMID: 25261218 Free PMC article.
-
MixGF: spectral probabilities for mixture spectra from more than one peptide.Mol Cell Proteomics. 2014 Dec;13(12):3688-97. doi: 10.1074/mcp.O113.037218. Epub 2014 Sep 15. Mol Cell Proteomics. 2014. PMID: 25225354 Free PMC article.
-
PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data.Nat Methods. 2017 Sep;14(9):903-908. doi: 10.1038/nmeth.4390. Epub 2017 Aug 7. Nat Methods. 2017. PMID: 28783153 Free PMC article.
References
-
- Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature biotechnology. 2001;19(3):242–247. - PubMed
-
- Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. Journal of proteome research. 2008;7(10):4598–4602. - PubMed
-
- Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5(11):976–989. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. - PubMed
-
- Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid communications in mass spectrometry. 2003;17(20):2310–2316. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
