

Whole-exome sequencing in a single proband reveals a mutation in the CHST8 gene in autosomal recessive peeling skin syndrome
- PMID: 22289416
- PMCID: PMC4362535
- DOI: 10.1016/j.ygeno.2012.01.005
Whole-exome sequencing in a single proband reveals a mutation in the CHST8 gene in autosomal recessive peeling skin syndrome
Abstract
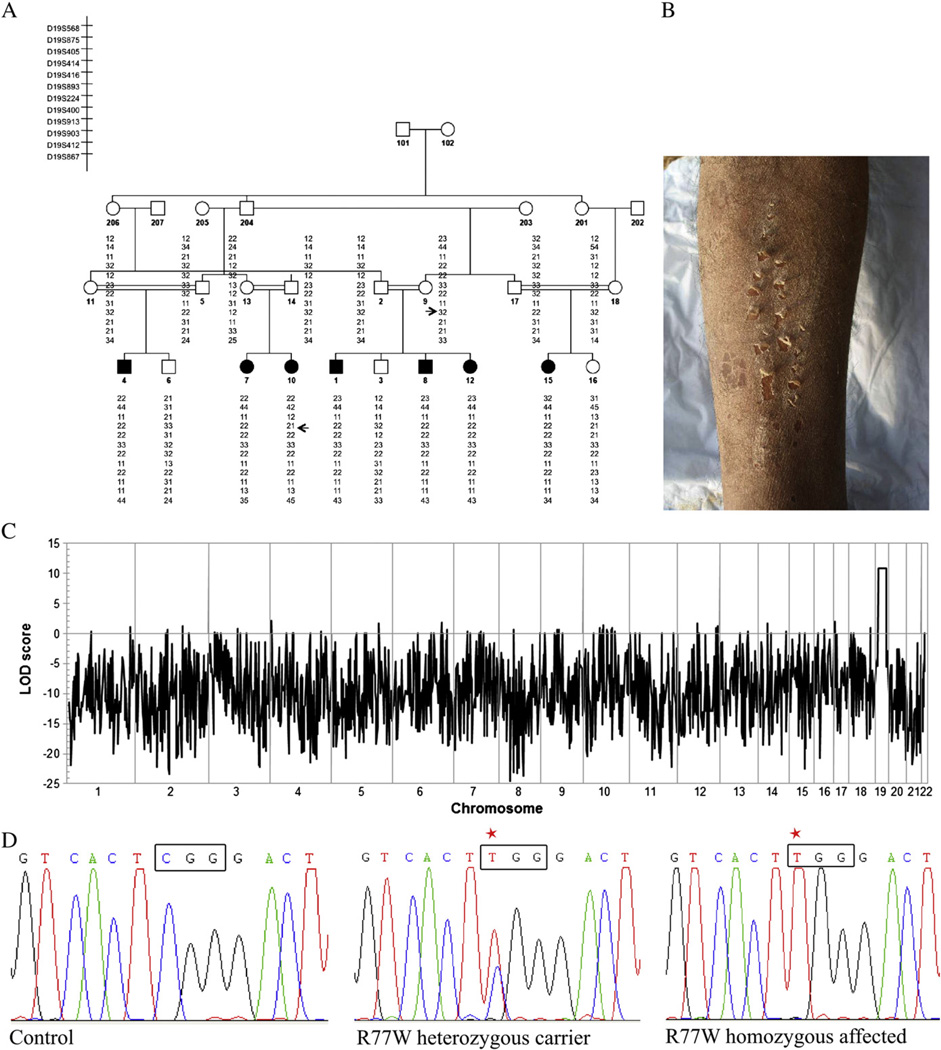
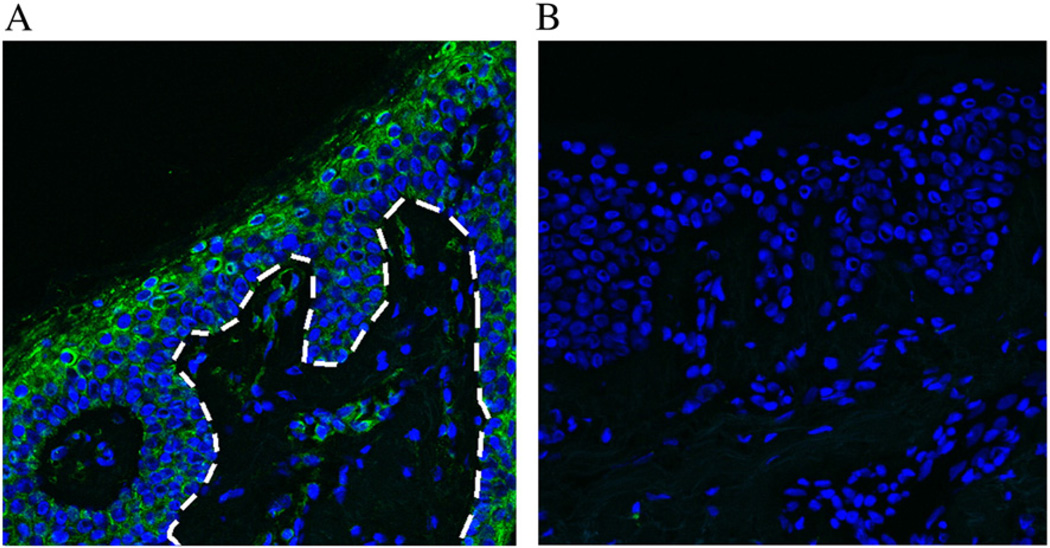
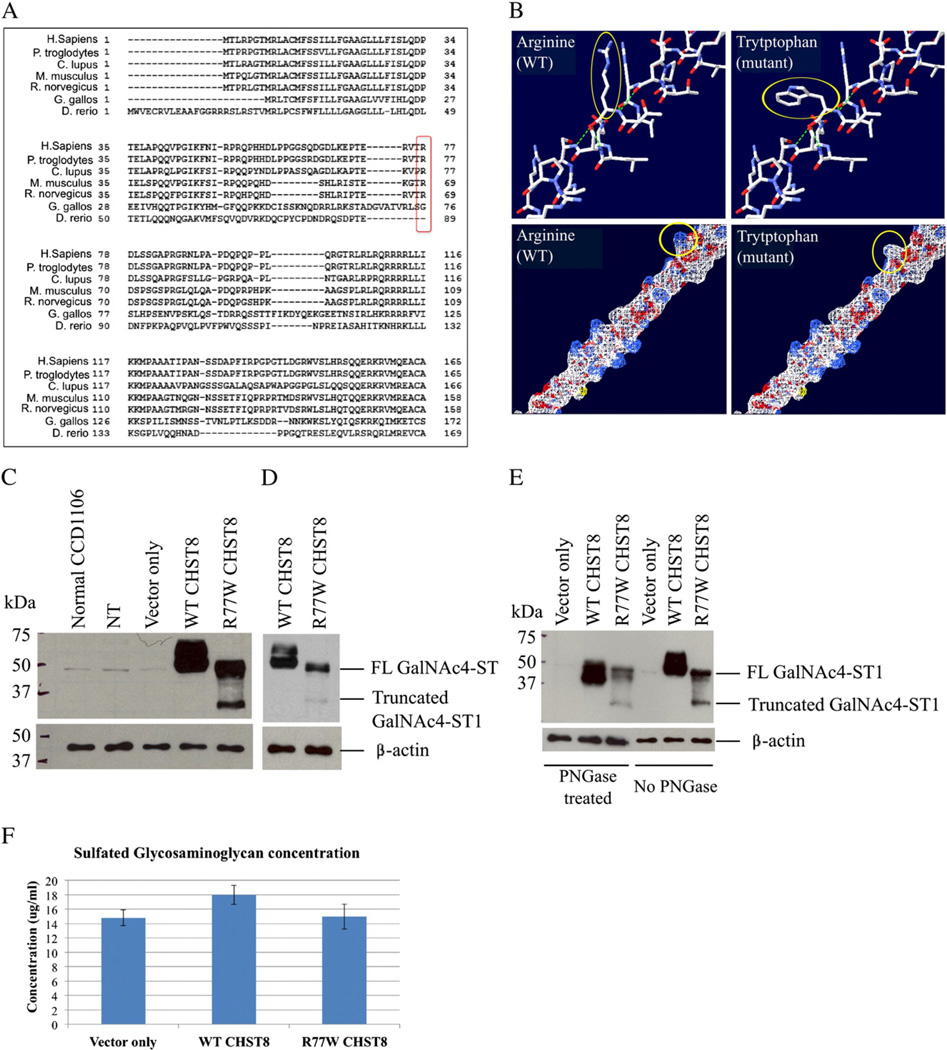
Generalized peeling skin syndrome (PSS) is an autosomal recessive genodermatosis characterized by lifelong, continuous shedding of the upper epidermis. Using whole-genome homozygozity mapping and whole-exome sequencing, we identified a novel homozygous missense mutation (c.229C>T, R77W) within the CHST8 gene, in a large consanguineous family with non-inflammatory PSS type A. CHST8 encodes a Golgi transmembrane N-acetylgalactosamine-4-O-sulfotransferase (GalNAc4-ST1), which we show by immunofluorescence staining to be expressed throughout normal epidermis. A colorimetric assay for total sulfated glycosaminoglycan (GAG) quantification, comparing human keratinocytes (CCD1106 KERTr) expressing wild type and mutant recombinant GalNAc4-ST1, revealed decreased levels of total sulfated GAGs in cells expressing mutant GalNAc4-ST1, suggesting loss of function. Western blotting revealed lower expression levels of mutant recombinant GalNAc4-ST1 compared to wild type, suggesting that accelerated degradation may result in loss of function, leading to PSS type A. This is the first report describing a mutation as the cause of PSS type A.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Janjua SA, Hussain I, Khachemoune A. Facial peeling skin syndrome: a case report and a brief review. Int. J. Dermatol. 2007;46:287–289. - PubMed
-
- Köse O, Safali M, Koç E, Arca E, Açikgöz G, Ozmen I, Yeniay Y. Peeling skin diseases: 21 cases from Turkey and a review of the literature. J. Eur. Acad. Dermatol. Venereol. 2011 [Ahead of print]. - PubMed
-
- Bowden PE. Peeling skin syndrome: genetic defects in late terminal differentiation of the epidermis. J. Investig. Dermatol. 2011;3:561–564. - PubMed
-
- Ishida-Yamamoto A, Igawa S, Kishibe M. Order and disorder in corneocyte adhesion. J. Dermatol. 2011;7:645–654. - PubMed
-
- Telem DF, Israeli S, Sarig O, Sprecher E. Inflammatory peeling skin syndrome caused a novel mutation in CDSN. Arch. Dermatol. Res. 2011 [Ahead of print]. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
