

Inference of population structure using dense haplotype data
- PMID: 22291602
- PMCID: PMC3266881
- DOI: 10.1371/journal.pgen.1002453
Inference of population structure using dense haplotype data
Abstract
The advent of genome-wide dense variation data provides an opportunity to investigate ancestry in unprecedented detail, but presents new statistical challenges. We propose a novel inference framework that aims to efficiently capture information on population structure provided by patterns of haplotype similarity. Each individual in a sample is considered in turn as a recipient, whose chromosomes are reconstructed using chunks of DNA donated by the other individuals. Results of this "chromosome painting" can be summarized as a "coancestry matrix," which directly reveals key information about ancestral relationships among individuals. If markers are viewed as independent, we show that this matrix almost completely captures the information used by both standard Principal Components Analysis (PCA) and model-based approaches such as STRUCTURE in a unified manner. Furthermore, when markers are in linkage disequilibrium, the matrix combines information across successive markers to increase the ability to discern fine-scale population structure using PCA. In parallel, we have developed an efficient model-based approach to identify discrete populations using this matrix, which offers advantages over PCA in terms of interpretability and over existing clustering algorithms in terms of speed, number of separable populations, and sensitivity to subtle population structure. We analyse Human Genome Diversity Panel data for 938 individuals and 641,000 markers, and we identify 226 populations reflecting differences on continental, regional, local, and family scales. We present multiple lines of evidence that, while many methods capture similar information among strongly differentiated groups, more subtle population structure in human populations is consistently present at a much finer level than currently available geographic labels and is only captured by the haplotype-based approach. The software used for this article, ChromoPainter and fineSTRUCTURE, is available from http://www.paintmychromosomes.com/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
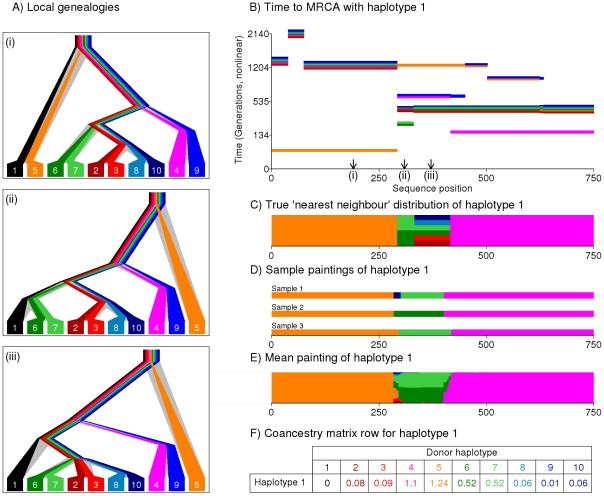

 regions and 20 individuals per population, showing
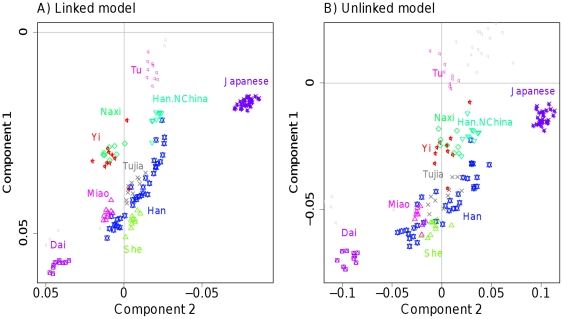
regions and 20 individuals per population, showing  for (bottom left) the unlinked model, and (top right) the linked model; note that the linked heatmap is slightly asymmetric. D) PCA applied to the dataset using Eigenstrat on the raw SNP data. E) PCA on the coancestry matrix assuming markers are unlinked and F) linked (see text for details).
for (bottom left) the unlinked model, and (top right) the linked model; note that the linked heatmap is slightly asymmetric. D) PCA applied to the dataset using Eigenstrat on the raw SNP data. E) PCA on the coancestry matrix assuming markers are unlinked and F) linked (see text for details).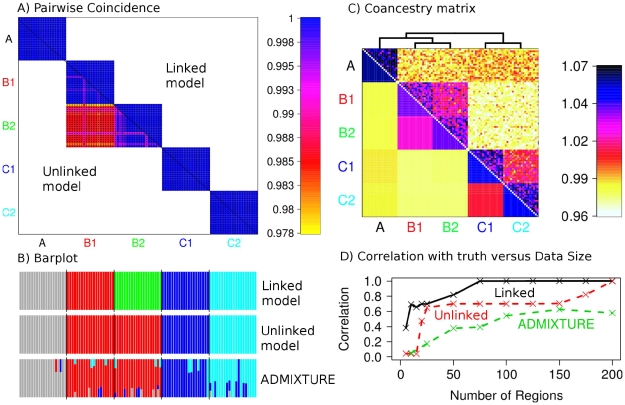
 ,
,  and
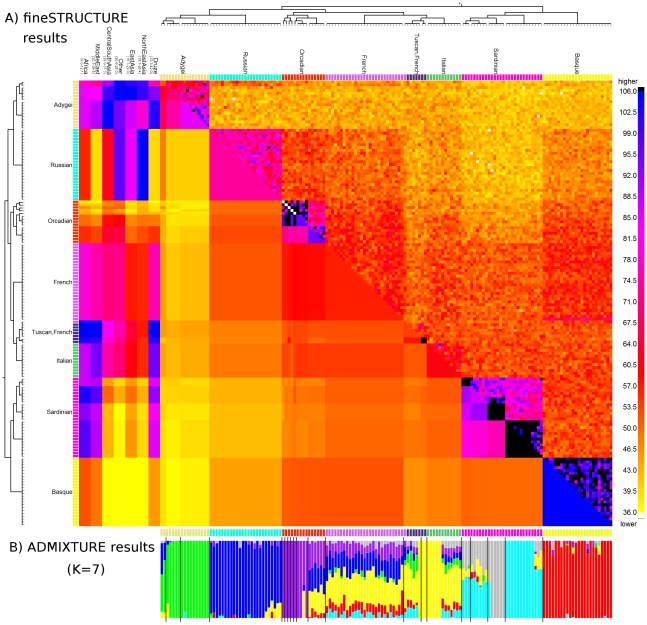
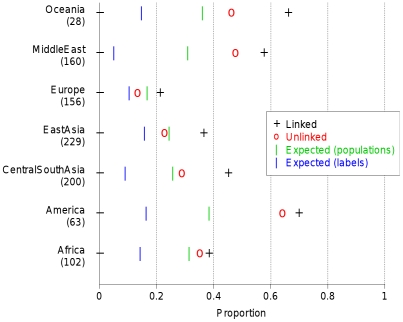
and  respectively). C) Aggregated coancestry matrix (bottom left, normalized to have row mean 1) for the linked model dataset (top right) rescaled from Figure 2C (also top right), shown with the inferred MAP tree (top). D) Correlation with the truth as a function of the number of 5 Mb data regions for fineSTRUCTURE linked and unlinked models, and ADMIXTURE on the same data.
respectively). C) Aggregated coancestry matrix (bottom left, normalized to have row mean 1) for the linked model dataset (top right) rescaled from Figure 2C (also top right), shown with the inferred MAP tree (top). D) Correlation with the truth as a function of the number of 5 Mb data regions for fineSTRUCTURE linked and unlinked models, and ADMIXTURE on the same data.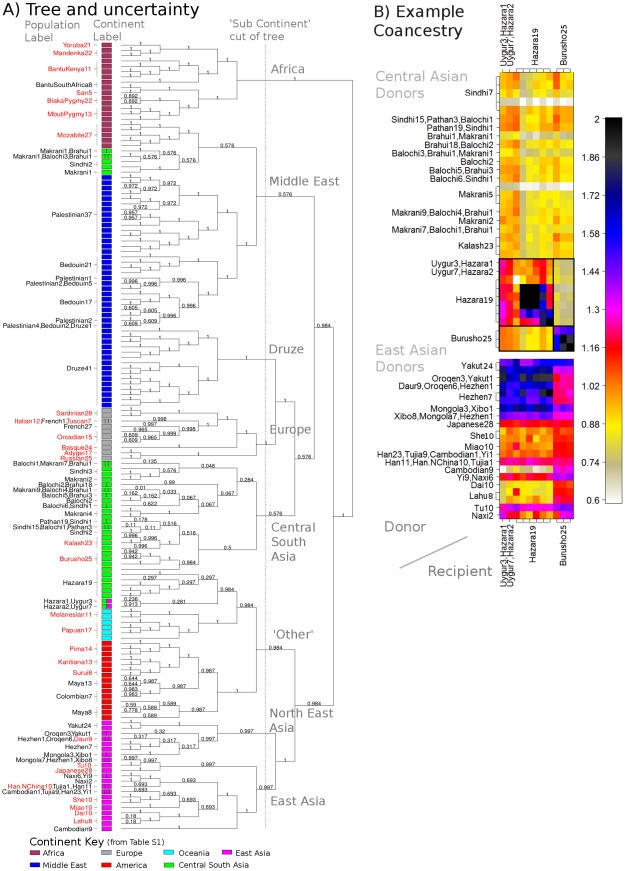
References
-
- Menozzi P, Piazza A, Cavalli-Sforza L. Synthetic maps of human gene frequencies in europeans. Science. 1978;201:786–792. - PubMed
-
- McVean G. A Genealogical Interpretation of Principal Components Analysis. PLoS Genet. 2009;5:e1000686. doi: 10.1371/journal.pgen.1000686. - DOI - PMC - PubMed
-
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 2006;38:904–909. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
