

Structure-function studies of DNA binding domain of response regulator KdpE reveals equal affinity interactions at DNA half-sites
- PMID: 22291906
- PMCID: PMC3264566
- DOI: 10.1371/journal.pone.0030102
Structure-function studies of DNA binding domain of response regulator KdpE reveals equal affinity interactions at DNA half-sites
Abstract
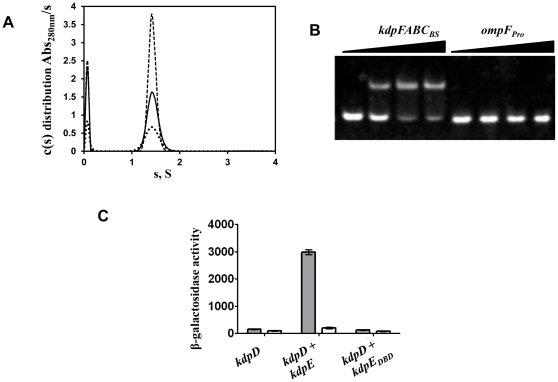
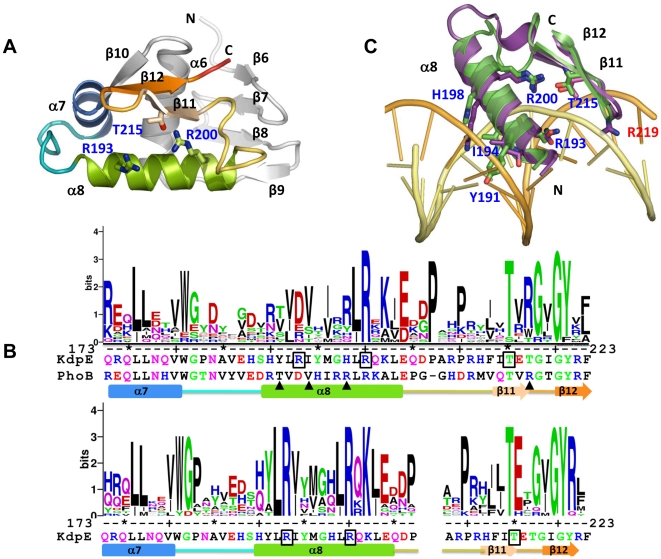
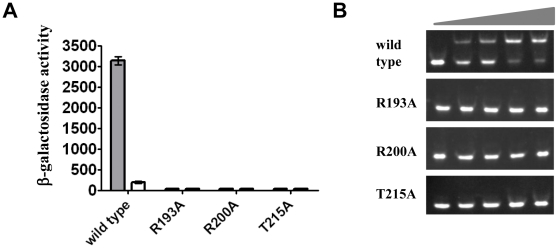
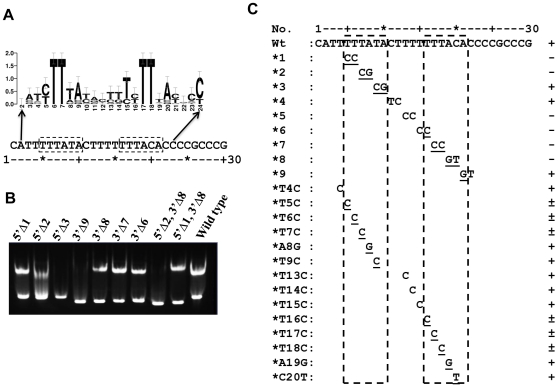
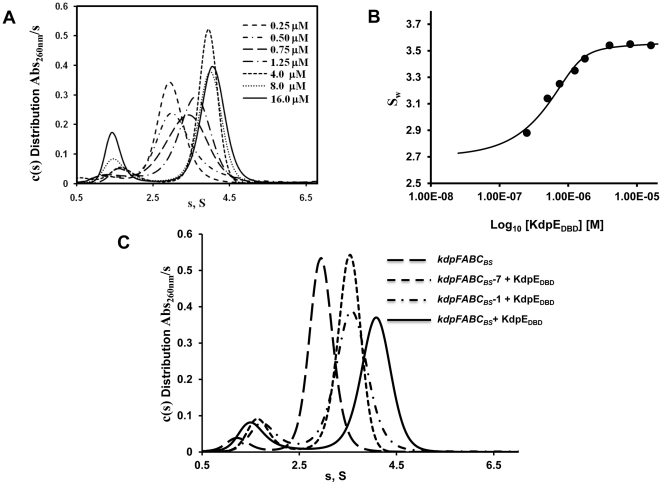
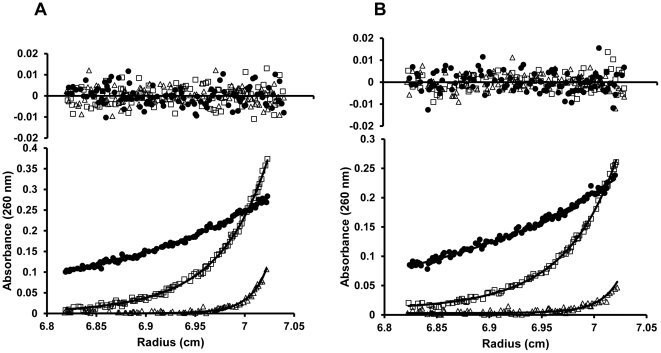
Expression of KdpFABC, a K(+) pump that restores osmotic balance, is controlled by binding of the response regulator KdpE to a specific DNA sequence (kdpFABC(BS)) via the winged helix-turn-helix type DNA binding domain (KdpE(DBD)). Exploration of E. coli KdpE(DBD) and kdpFABC(BS) interaction resulted in the identification of two conserved, AT-rich 6 bp direct repeats that form half-sites. Despite binding to these half-sites, KdpE(DBD) was incapable of promoting gene expression in vivo. Structure-function studies guided by our 2.5 Å X-ray structure of KdpE(DBD) revealed the importance of residues R193 and R200 in the α-8 DNA recognition helix and T215 in the wing region for DNA binding. Mutation of these residues renders KdpE incapable of inducing expression of the kdpFABC operon. Detailed biophysical analysis of interactions using analytical ultracentrifugation revealed a 2∶1 stoichiometry of protein to DNA with dissociation constants of 200±100 and 350±100 nM at half-sites. Inactivation of one half-site does not influence binding at the other, indicating that KdpE(DBD) binds independently to the half-sites with approximately equal affinity and no discernable cooperativity. To our knowledge, these data are the first to describe in quantitative terms the binding at half-sites under equilibrium conditions for a member of the ubiquitous OmpR/PhoB family of proteins.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
