

Signaling in innate immunity and inflammation
- PMID: 22296764
- PMCID: PMC3282411
- DOI: 10.1101/cshperspect.a006049
Signaling in innate immunity and inflammation
Abstract
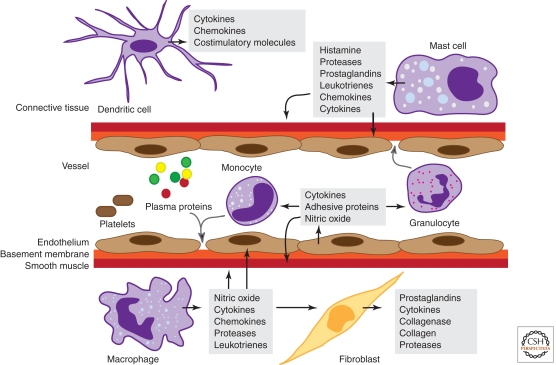
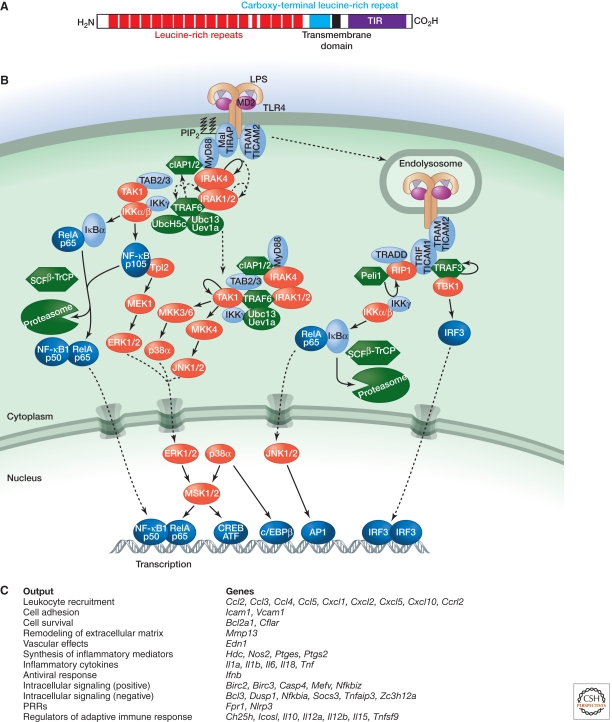
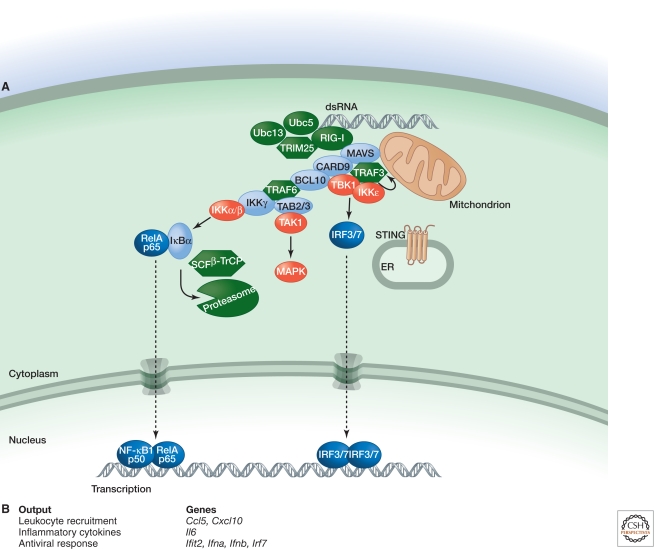
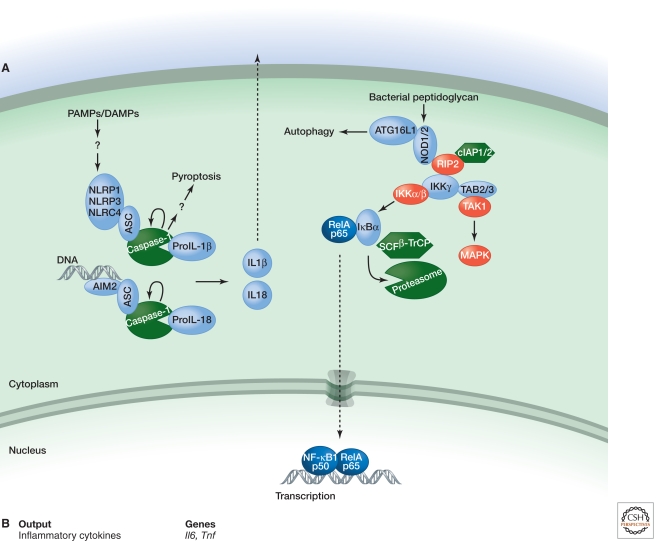
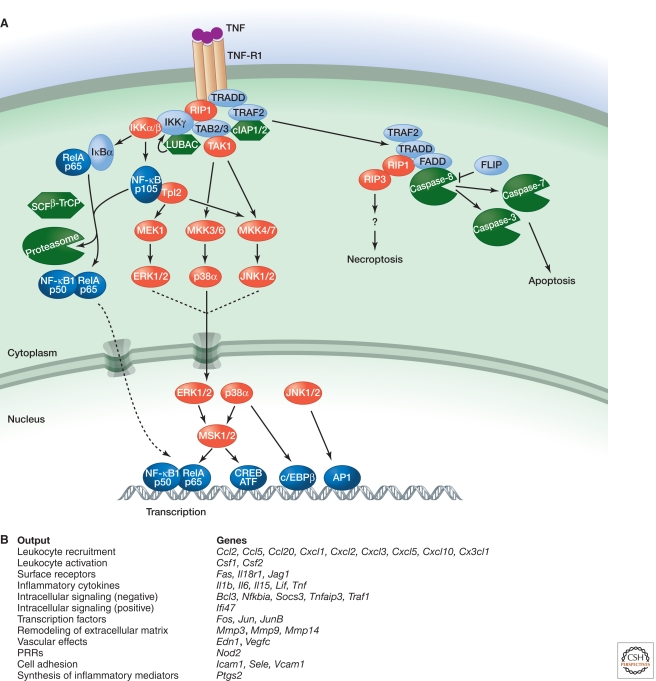
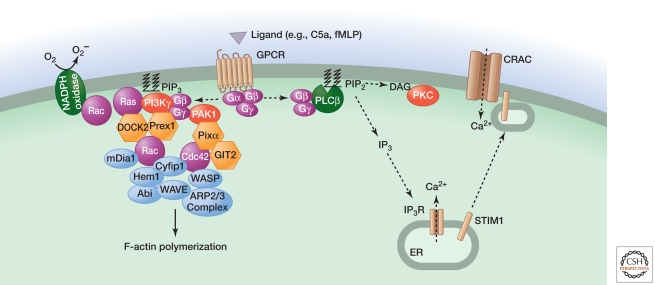
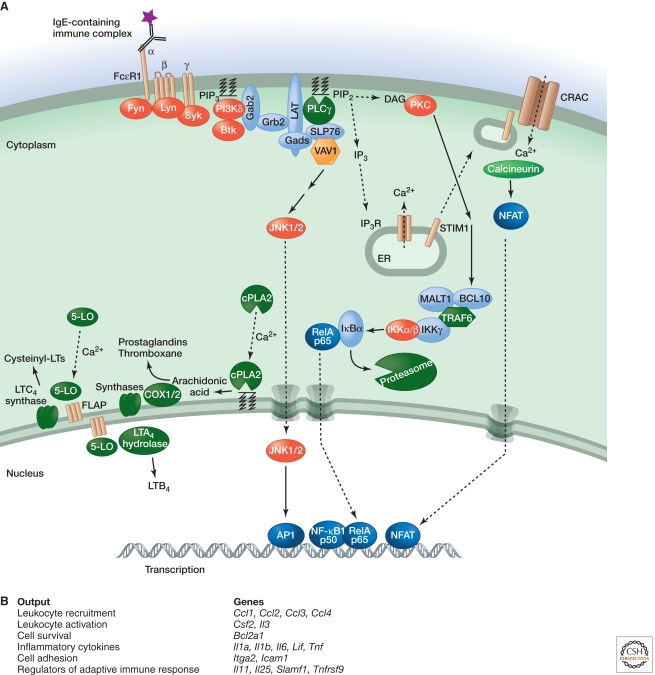
Inflammation is triggered when innate immune cells detect infection or tissue injury. Surveillance mechanisms involve pattern recognition receptors (PRRs) on the cell surface and in the cytoplasm. Most PRRs respond to pathogen-associated molecular patterns (PAMPs) or host-derived damage-associated molecular patterns (DAMPs) by triggering activation of NF-κB, AP1, CREB, c/EBP, and IRF transcription factors. Induction of genes encoding enzymes, chemokines, cytokines, adhesion molecules, and regulators of the extracellular matrix promotes the recruitment and activation of leukocytes, which are critical for eliminating foreign particles and host debris. A subset of PRRs activates the protease caspase-1, which causes maturation of the cytokines IL1β and IL18. Cell adhesion molecules and chemokines facilitate leukocyte extravasation from the circulation to the affected site, the chemokines stimulating G-protein-coupled receptors (GPCRs). Binding initiates signals that regulate leukocyte motility and effector functions. Other triggers of inflammation include allergens, which form antibody complexes that stimulate Fc receptors on mast cells. Although the role of inflammation is to resolve infection and injury, increasing evidence indicates that chronic inflammation is a risk factor for cancer.
Figures
References
-
- Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T 2008. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol 9: 81–88 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous