

Blood-brain barrier pathophysiology in traumatic brain injury
- PMID: 22299022
- PMCID: PMC3268209
- DOI: 10.1007/s12975-011-0125-x
Blood-brain barrier pathophysiology in traumatic brain injury
Abstract
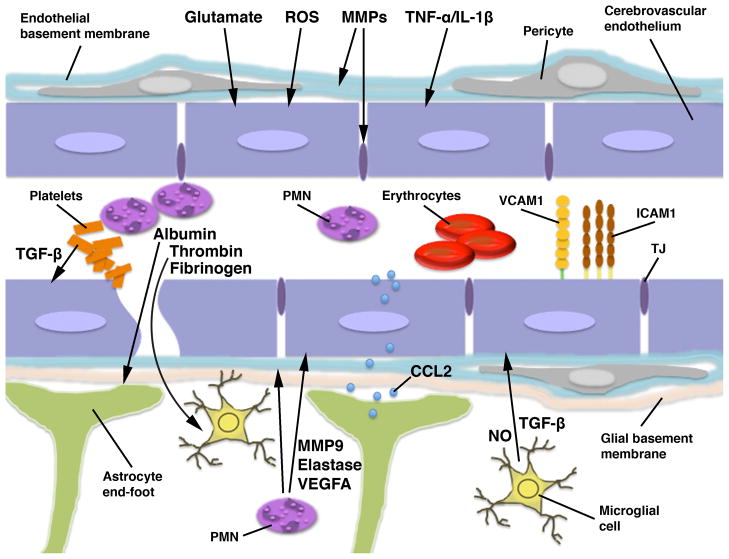
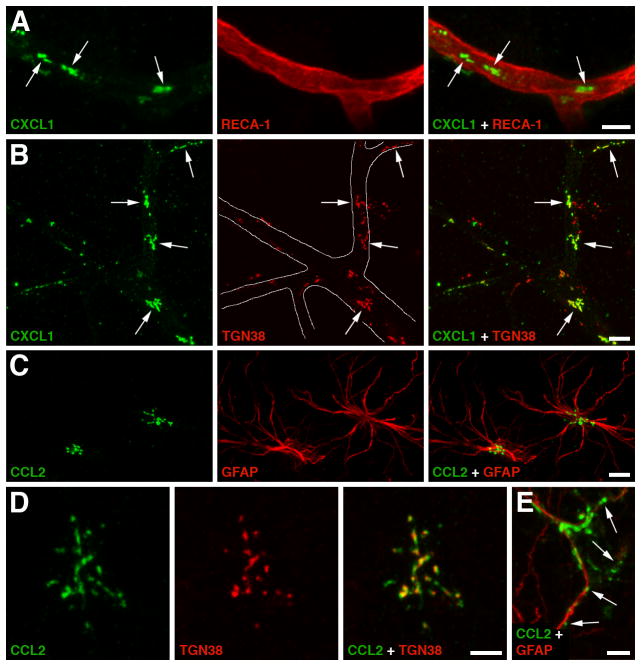
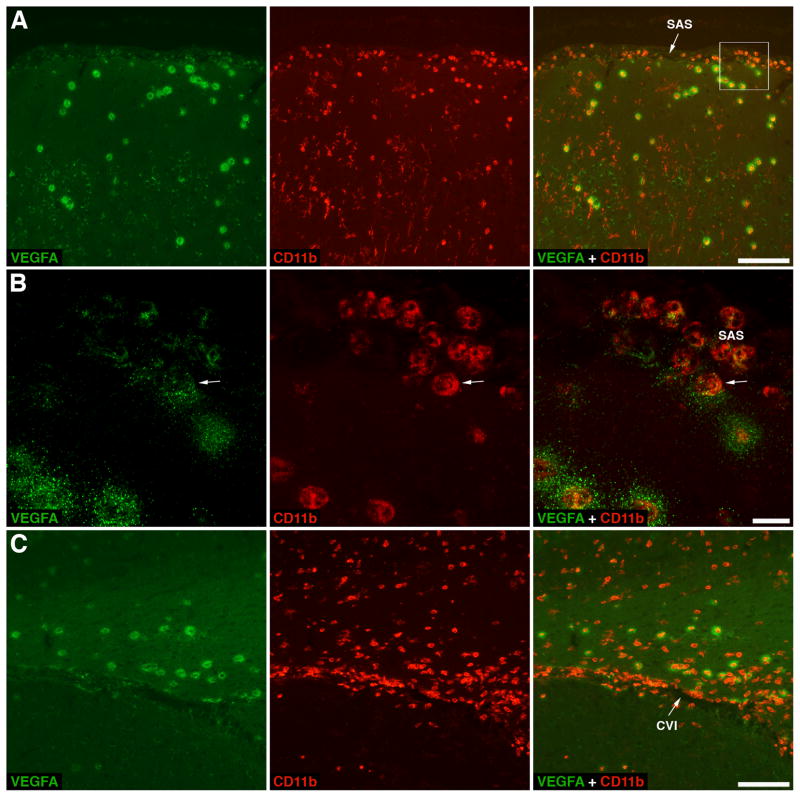
The blood-brain barrier (BBB) is formed by tightly connected cerebrovascular endothelial cells, but its normal function also depends on paracrine interactions between the brain endothelium and closely located glia. There is a growing consensus that brain injury, whether it is ischemic, hemorrhagic, or traumatic, leads to dysfunction of the BBB. Changes in BBB function observed after injury are thought to contribute to the loss of neural tissue and to affect the response to neuroprotective drugs. New discoveries suggest that considering the entire gliovascular unit, rather than the BBB alone, will expand our understanding of the cellular and molecular responses to traumatic brain injury (TBI). This review will address the BBB breakdown in TBI, the role of blood-borne factors in affecting the function of the gliovascular unit, changes in BBB permeability and post-traumatic edema formation, and the major pathophysiological factors associated with TBI that may contribute to post-traumatic dysfunction of the BBB. The key role of neuroinflammation and the possible effect of injury on transport mechanisms at the BBB will also be described. Finally, the potential role of the BBB as a target for therapeutic intervention through restoration of normal BBB function after injury and/or by harnessing the cerebrovascular endothelium to produce neurotrophic growth factors will be discussed.
Conflict of interest statement
The authors have no financial and/or competing interests to disclose.
Figures
References
-
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. - PubMed
-
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335(1):75–96. - PubMed
-
- Lassmann H, Zimprich F, Vass K, Hickey WF. Microglial cells are a component of the perivascular glia limitans. J Neurosci Res. 1991;28(2):236–43. - PubMed
-
- Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, Engelhardt B, Grammas P, Nedergaard M, Nutt J, et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7(1):84–96. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
