

Effects of sequence variation on differential allelic transcription factor occupancy and gene expression
- PMID: 22300769
- PMCID: PMC3337432
- DOI: 10.1101/gr.131201.111
Effects of sequence variation on differential allelic transcription factor occupancy and gene expression
Abstract
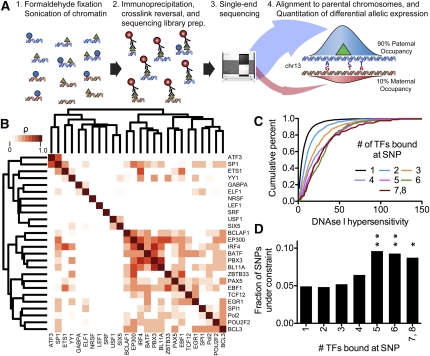
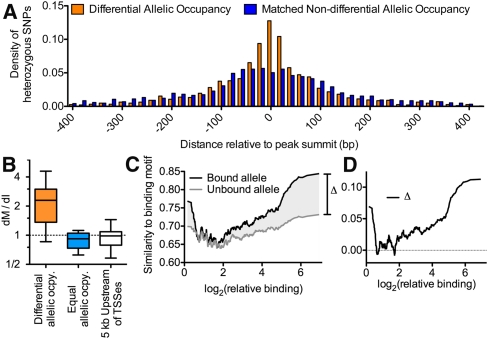
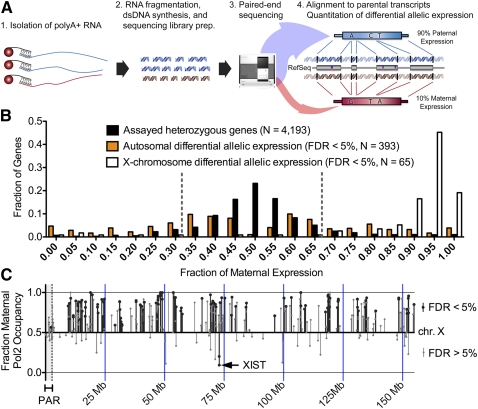
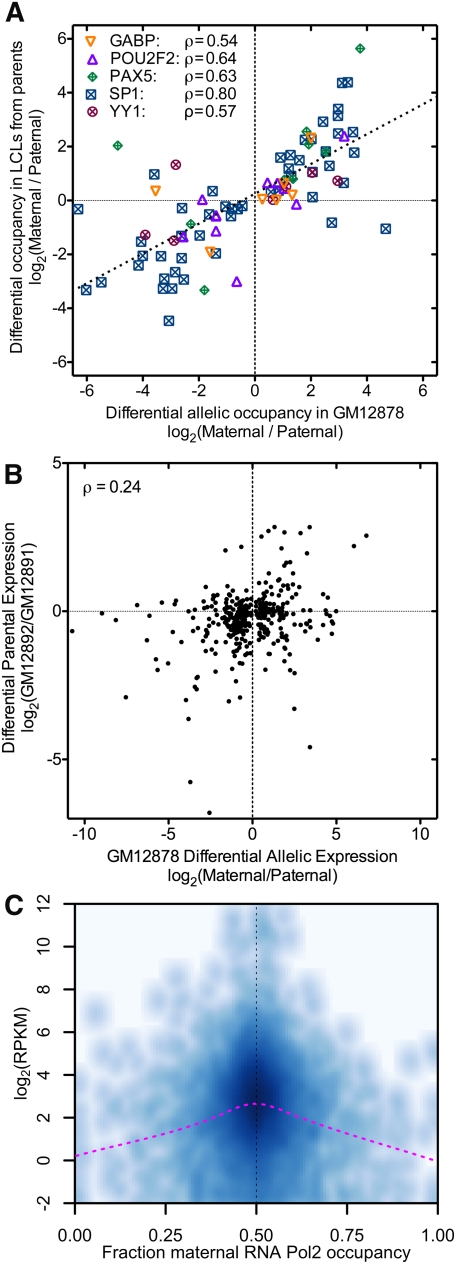
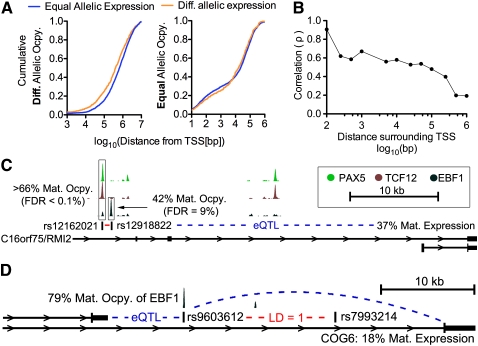
A complex interplay between transcription factors (TFs) and the genome regulates transcription. However, connecting variation in genome sequence with variation in TF binding and gene expression is challenging due to environmental differences between individuals and cell types. To address this problem, we measured genome-wide differential allelic occupancy of 24 TFs and EP300 in a human lymphoblastoid cell line GM12878. Overall, 5% of human TF binding sites have an allelic imbalance in occupancy. At many sites, TFs clustered in TF-binding hubs on the same homolog in especially open chromatin. While genetic variation in core TF binding motifs generally resulted in large allelic differences in TF occupancy, most allelic differences in occupancy were subtle and associated with disruption of weak or noncanonical motifs. We also measured genome-wide differential allelic expression of genes with and without heterozygous exonic variants in the same cells. We found that genes with differential allelic expression were overall less expressed both in GM12878 cells and in unrelated human cell lines. Comparing TF occupancy with expression, we found strong association between allelic occupancy and expression within 100 bp of transcription start sites (TSSs), and weak association up to 100 kb from TSSs. Sites of differential allelic occupancy were significantly enriched for variants associated with disease, particularly autoimmune disease, suggesting that allelic differences in TF occupancy give functional insights into intergenic variants associated with disease. Our results have the potential to increase the power and interpretability of association studies by targeting functional intergenic variants in addition to protein coding sequences.
Figures
Comment in
-
Functional genomics: Complexities of occupancy and sequence.Nat Rev Genet. 2012 Apr 12;13(5):297. doi: 10.1038/nrg3219. Nat Rev Genet. 2012. PMID: 22495436 No abstract available.
References
-
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300
-
- Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF 1991. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349: 38–44 - PubMed
-
- Carrel L, Willard HF 2005. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434: 400–404 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous