

Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death
- PMID: 22302006
- PMCID: PMC3337840
- DOI: 10.4161/auto.18658
Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death
Abstract
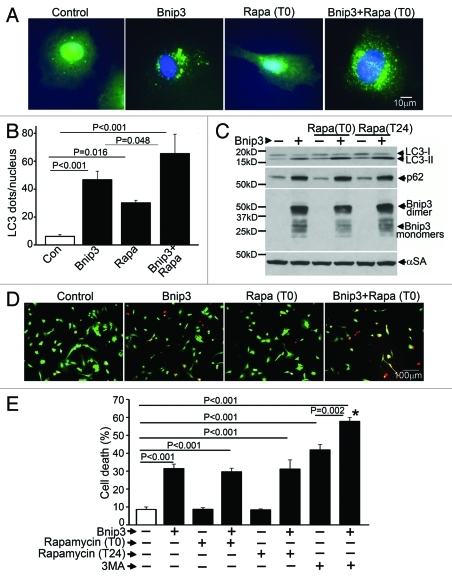
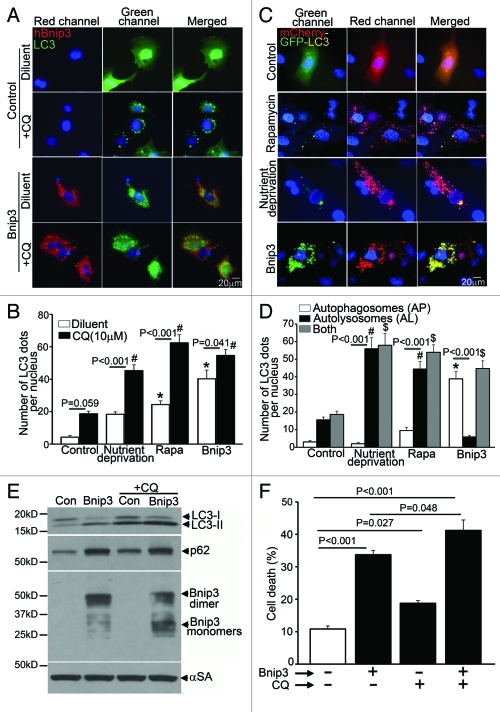
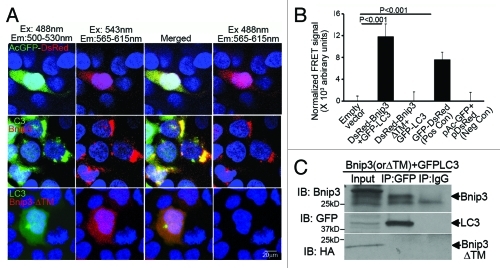
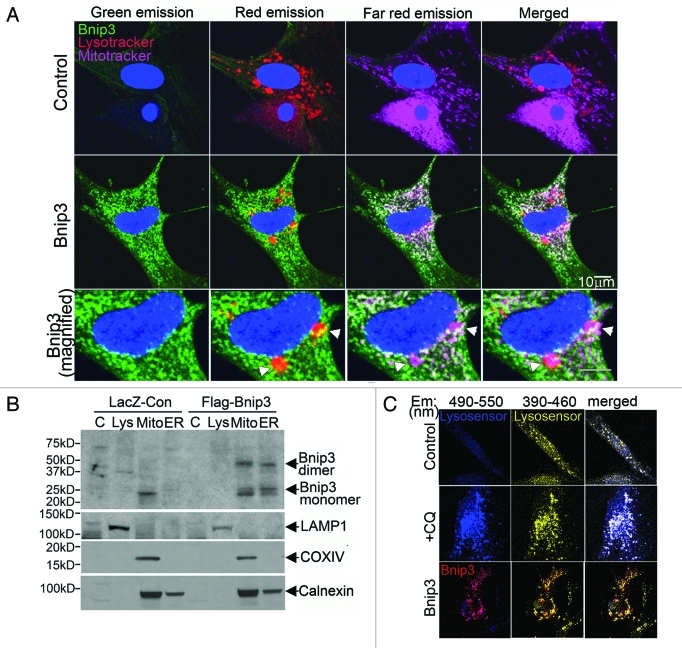
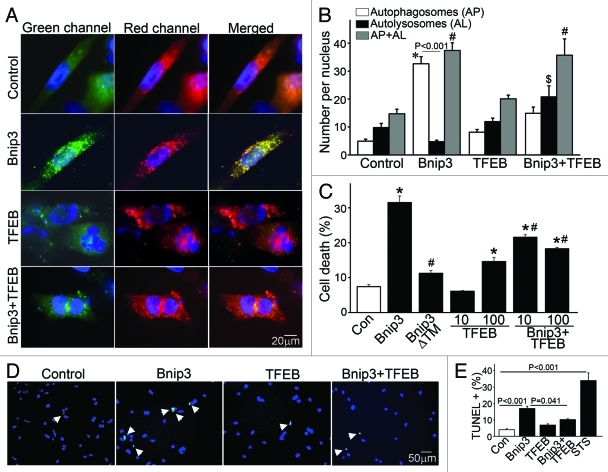
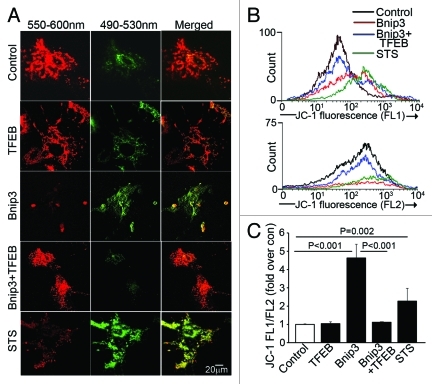
Hypoxia-inducible pro-death protein BNIP3 (BCL-2/adenovirus E1B 19-kDa interacting protein 3), provokes mitochondrial permeabilization causing cardiomyocyte death in ischemia-reperfusion injury. Inhibition of autophagy accelerates BNIP3-induced cell death, by preventing removal of damaged mitochondria. We tested the hypothesis that stimulating autophagy will attenuate BNIP3-induced cardiomyocyte death. Neonatal rat cardiac myocytes (NRCMs) were adenovirally transduced with BNIP3 (or LacZ as control; at multiplicity of infection = 100); and autophagy was stimulated with rapamycin (100 nM). Cell death was assessed at 48 h. BNIP3 expression increased autophagosome abundance 8-fold and caused a 3.6-fold increase in cardiomyocyte death as compared with control. Rapamycin treatment of BNIP3-expressing cells led to further increase in autophagosome number without affecting cell death. BNIP3 expression led to accumulation of autophagosome-bound LC3-II and p62, and an increase in autophagosomes, but not autolysosomes (assessed with dual fluorescent mCherry-GFP-LC3 expression). BNIP3, but not the transmembrane deletion variant, interacted with LC3 and colocalized with mitochondria and lysosomes. However, BNIP3 did not target to lysosomes by subcellular fractionation, provoke lysosome permeabilization or alter lysosome pH. Rather, BNIP3-induced autophagy caused a decline in lysosome numbers with decreased expression of the lysosomal protein LAMP-1, indicating lysosome consumption and consequent autophagosome accumulation. Forced expression of transcription factor EB (TFEB) in BNIP3-expressing cells increased lysosome numbers, decreased autophagosomes and increased autolysosomes, prevented p62 accumulation, removed depolarized mitochondria and attenuated BNIP3-induced death. We conclude that BNIP3 expression induced autophagosome accumulation with lysosome consumption in cardiomyocytes. Forced expression of TFEB, a lysosomal biogenesis factor, restored autophagosome processing and attenuated BNIP3-induced cell death.
Figures

Similar articles
-
Regulation of the transcription factor EB-PGC1α axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress.Mol Cell Biol. 2015 Mar;35(6):956-76. doi: 10.1128/MCB.01091-14. Epub 2015 Jan 5. Mol Cell Biol. 2015. PMID: 25561470 Free PMC article.
-
Downregulation of LAPTM4B Contributes to the Impairment of the Autophagic Flux via Unopposed Activation of mTORC1 Signaling During Myocardial Ischemia/Reperfusion Injury.Circ Res. 2020 Sep 11;127(7):e148-e165. doi: 10.1161/CIRCRESAHA.119.316388. Epub 2020 Jul 22. Circ Res. 2020. PMID: 32693673
-
Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury.Autophagy. 2015;11(9):1537-60. doi: 10.1080/15548627.2015.1063768. Autophagy. 2015. PMID: 26103523 Free PMC article.
-
A unique pathway of cardiac myocyte death caused by hypoxia-acidosis.J Exp Biol. 2004 Aug;207(Pt 18):3189-200. doi: 10.1242/jeb.01109. J Exp Biol. 2004. PMID: 15299040 Review.
-
BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions.Oncogene. 2008 Dec;27 Suppl 1(Suppl 1):S114-27. doi: 10.1038/onc.2009.49. Oncogene. 2008. PMID: 19641497 Free PMC article. Review.
Cited by
-
Metformin alleviates bone loss in ovariectomized mice through inhibition of autophagy of osteoclast precursors mediated by E2F1.Cell Commun Signal. 2022 Oct 25;20(1):165. doi: 10.1186/s12964-022-00966-5. Cell Commun Signal. 2022. PMID: 36284303 Free PMC article.
-
Eicosapentaenoic acid attenuated oxidative stress-induced cardiomyoblast apoptosis by activating adaptive autophagy.Eur J Nutr. 2014;53(2):541-7. doi: 10.1007/s00394-013-0562-2. Epub 2013 Jul 26. Eur J Nutr. 2014. PMID: 23887854
-
Propofol Inhibits HeLa Cells by Impairing Autophagic Flux via AMP-Activated Protein Kinase (AMPK) Activation and Endoplasmic Reticulum Stress Regulated by Calcium.Med Sci Monit. 2018 Apr 18;24:2339-2349. doi: 10.12659/msm.909144. Med Sci Monit. 2018. PMID: 29667627 Free PMC article.
-
IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions.Sci Signal. 2015 Jun 23;8(382):ra62. doi: 10.1126/scisignal.aaa0341. Sci Signal. 2015. PMID: 26106220 Free PMC article.
-
p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles.Int J Mol Sci. 2020 Jul 16;21(14):5029. doi: 10.3390/ijms21145029. Int J Mol Sci. 2020. PMID: 32708719 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous