

Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort
- PMID: 22302274
- PMCID: PMC3752368
- DOI: 10.1007/s00415-011-6397-y
Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort
Abstract
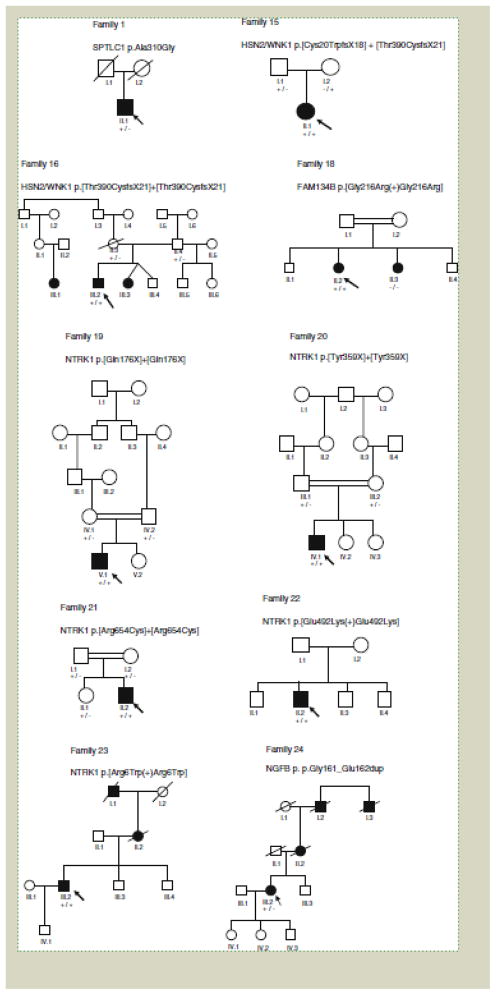
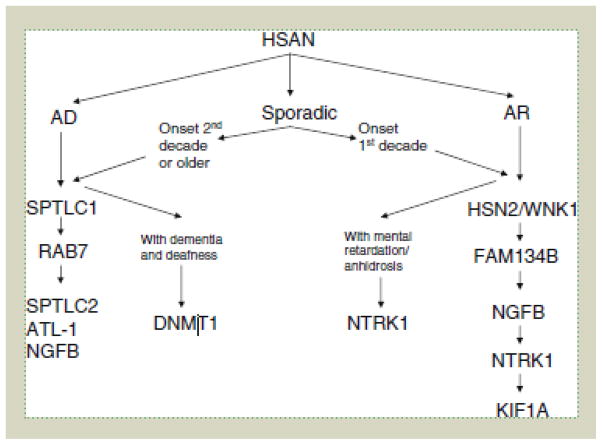
The hereditary sensory and autonomic neuropathies (HSAN, also known as the hereditary sensory neuropathies) are a clinically and genetically heterogeneous group of disorders, characterised by a progressive sensory neuropathy often complicated by ulcers and amputations, with variable motor and autonomic involvement. To date, mutations in twelve genes have been identified as causing HSAN. To study the frequency of mutations in these genes and the associated phenotypes, we screened 140 index patients in our inherited neuropathy cohort with a clinical diagnosis of HSAN for mutations in the coding regions of SPTLC1, RAB7, WNK1/HSN2, FAM134B, NTRK1 (TRKA) and NGFB. We identified 25 index patients with mutations in six genes associated with HSAN (SPTLC1, RAB7, WNK1/HSN2, FAM134B, NTRK1 and NGFB); 20 of which appear to be pathogenic giving an overall mutation frequency of 14.3%. Mutations in the known genes for HSAN are rare suggesting that further HSAN genes are yet to be identified. The p.Cys133Trp mutation in SPTLC1 is the most common cause of HSAN in the UK population and should be screened first in all patients with sporadic or autosomal dominant HSAN.
Conflict of interest statement
Figures
References
-
- Dyck PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral neuropathy. W.B Saunders; Philadelphia: 1993. pp. 1065–1093.
-
- Eichler FS, Hornemann T, McCampbell A, Kuljis D, Penno A, Vardeh D, Tamrazian E, Garofalo K, Lee H-J, Kini L, Selig M, Frosch M, Gable K, von Eckardstein A, Woolf CJ, Guan G, Harmon JM, Dunn TM, Brown RH., Jr Overexpression of the wild-type SPT1 subunit lowers deoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci. 2009;29:14646–14651. - PMC - PubMed
-
- Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, Solders G, Holmgren G, Holmberg D, Holmberg M. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. 2004;13:799–805. - PubMed
-
- Fitzgibbon GJ, Kingston H, Needham M, Gaunt L. Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain. Dev Med Child Neurol. 2009;51:833–837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
