

Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system
- PMID: 22302938
- PMCID: PMC3278893
- DOI: 10.1101/gad.180406.111
Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system
Abstract
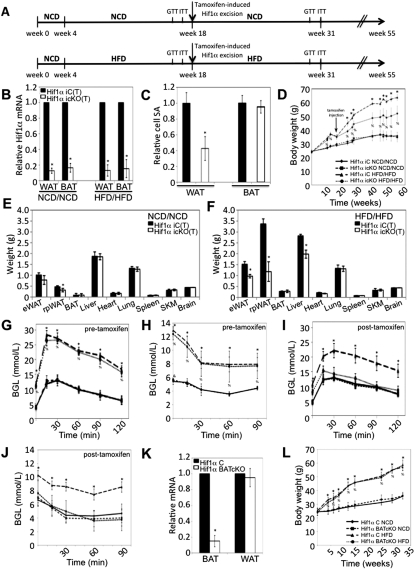
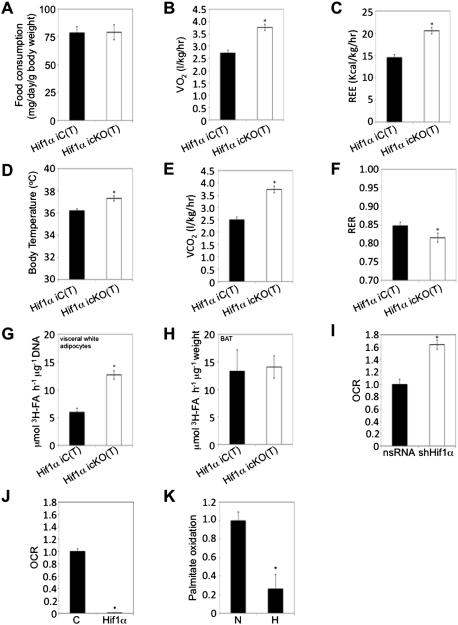
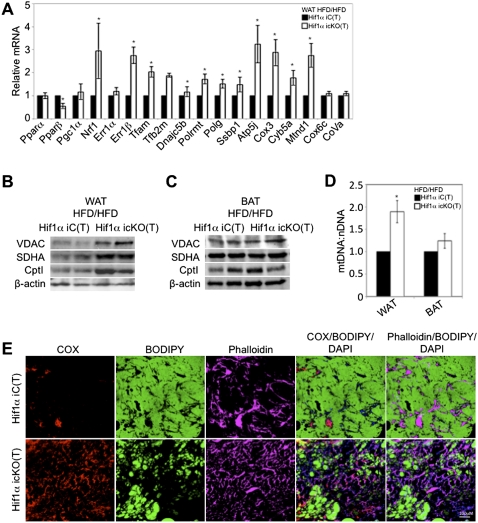
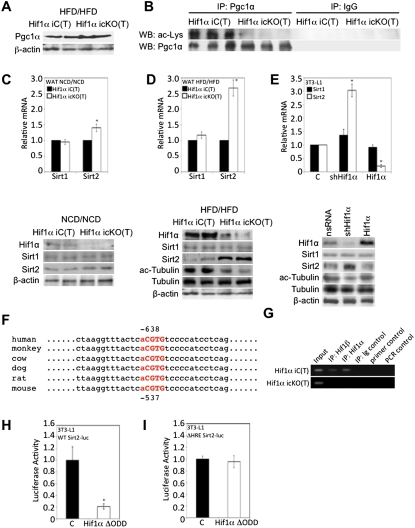
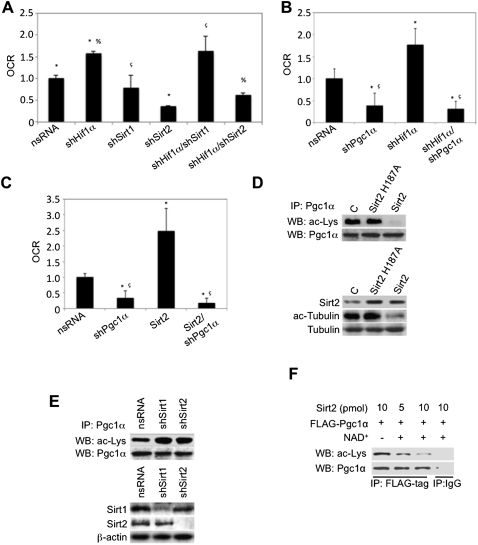
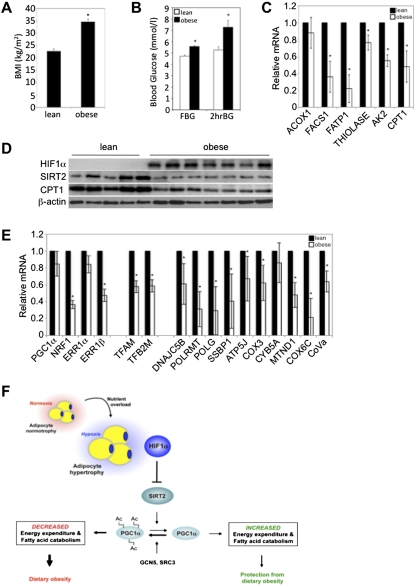
Dietary obesity is a major factor in the development of type 2 diabetes and is associated with intra-adipose tissue hypoxia and activation of hypoxia-inducible factor 1α (HIF1α). Here we report that, in mice, Hif1α activation in visceral white adipocytes is critical to maintain dietary obesity and associated pathologies, including glucose intolerance, insulin resistance, and cardiomyopathy. This function of Hif1α is linked to its capacity to suppress β-oxidation, in part, through transcriptional repression of sirtuin 2 (Sirt2) NAD(+)-dependent deacetylase. Reduced Sirt2 function directly translates into diminished deacetylation of PPARγ coactivator 1α (Pgc1α) and expression of β-oxidation and mitochondrial genes. Importantly, visceral adipose tissue from human obese subjects is characterized by high levels of HIF1α and low levels of SIRT2. Thus, by negatively regulating the Sirt2-Pgc1α regulatory axis, Hif1α negates adipocyte-intrinsic pathways of fatty acid catabolism, thereby creating a metabolic state supporting the development of obesity.
Figures
References
-
- Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Scholmerich J, Bollheimer LC 2006. Defining high-fat-diet rat models: Metabolic and molecular effects of different fat types. J Mol Endocrinol 36: 485–501 - PubMed
-
- Cinti S 2009. Transdifferentiation properties of adipocytes in the adipose organ. Am J Physiol 297: E977–E986 doi: 10.1152/ajpendo.00183.2009 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases