

Whole genome sequences of a male and female supercentenarian, ages greater than 114 years
- PMID: 22303384
- PMCID: PMC3262222
- DOI: 10.3389/fgene.2011.00090
Whole genome sequences of a male and female supercentenarian, ages greater than 114 years
Abstract
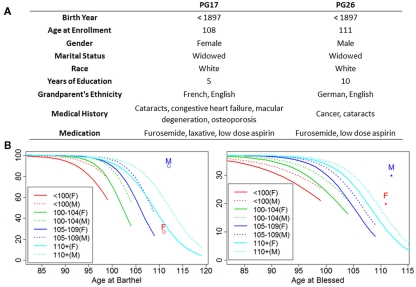
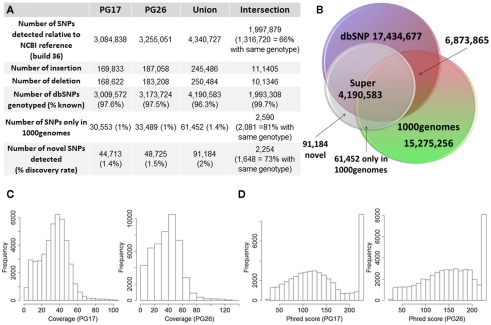
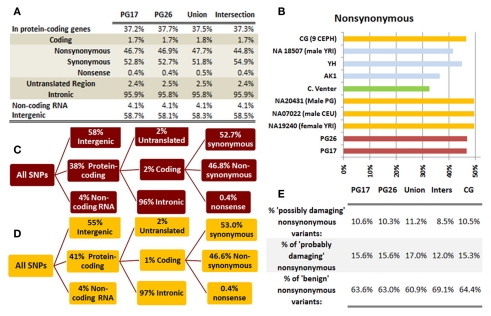
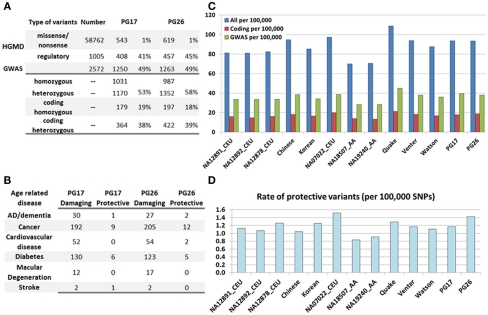
Supercentenarians (age 110+ years old) generally delay or escape age-related diseases and disability well beyond the age of 100 and this exceptional survival is likely to be influenced by a genetic predisposition that includes both common and rare genetic variants. In this report, we describe the complete genomic sequences of male and female supercentenarians, both age >114 years old. We show that: (1) the sequence variant spectrum of these two individuals' DNA sequences is largely comparable to existing non-supercentenarian genomes; (2) the two individuals do not appear to carry most of the well-established human longevity enabling variants already reported in the literature; (3) they have a comparable number of known disease-associated variants relative to most human genomes sequenced to-date; (4) approximately 1% of the variants these individuals possess are novel and may point to new genes involved in exceptional longevity; and (5) both individuals are enriched for coding variants near longevity-associated variants that we discovered through a large genome-wide association study. These analyses suggest that there are both common and rare longevity-associated variants that may counter the effects of disease-predisposing variants and extend lifespan. The continued analysis of the genomes of these and other rare individuals who have survived to extremely old ages should provide insight into the processes that contribute to the maintenance of health during extreme aging.
Keywords: aging; centenarian; genetics; longevity; supercentenarian; whole genome sequence.
Figures
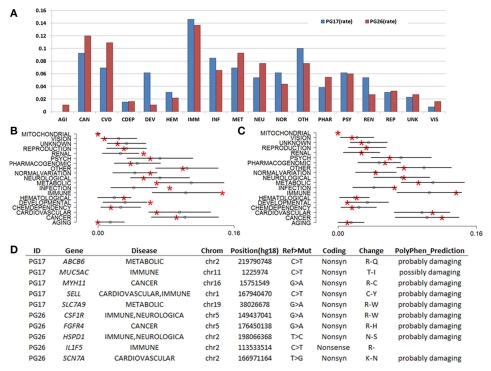
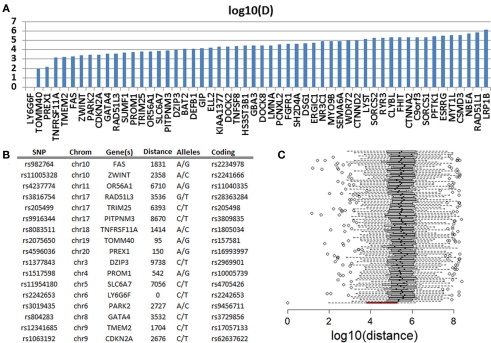
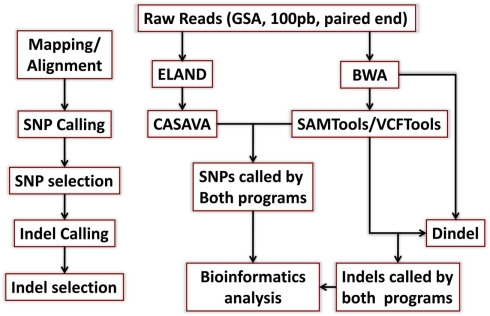
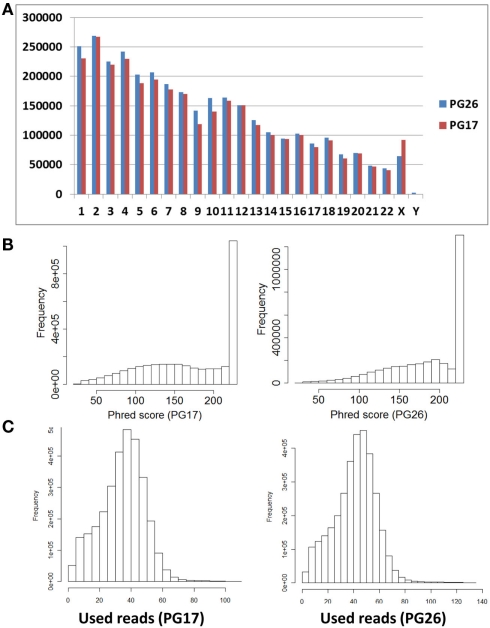

References
-
- Arai Y., Takayama M., Gondo Y., Inagaki H., Yamamura K., Nakazawa S., Kojima T., Ebihara Y., Shimizu K., Masui Y., Kitagawa K., Takebayashi T., Hirose N. (2008). Adipose endocrine function, insulin-like growth factor-1 axis, and exceptional survival beyond 100 years of age. J. Gerontol. A Biol. Sci. Med. Sci. 63, 1209–1218 10.1093/gerona/63.11.1209 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases