

Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells
- PMID: 22303462
- PMCID: PMC3267756
- DOI: 10.1371/journal.pone.0030841
Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells
Abstract
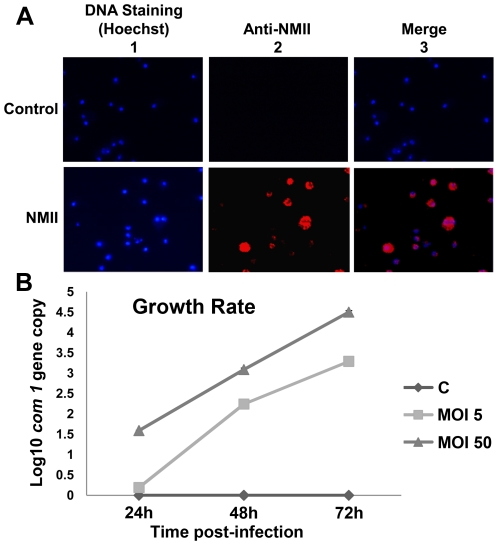
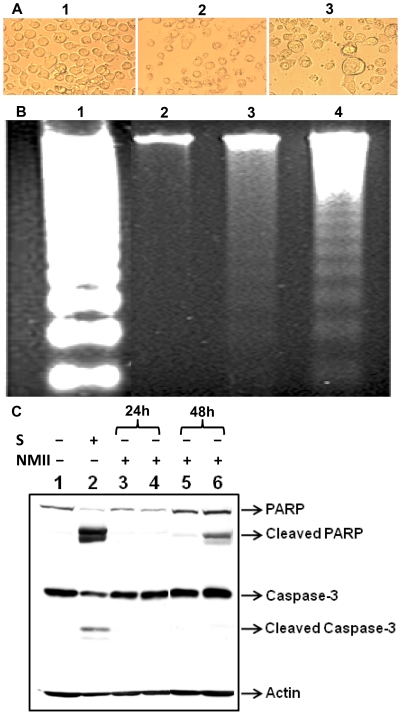
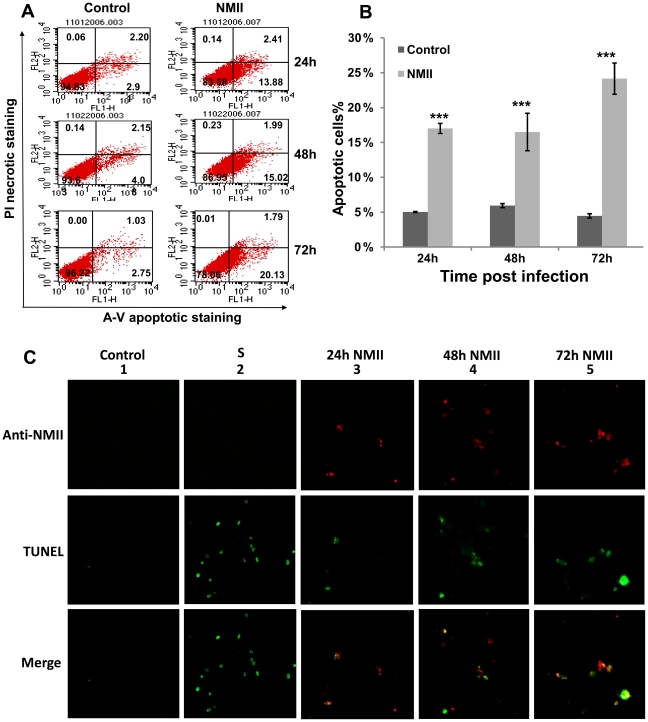
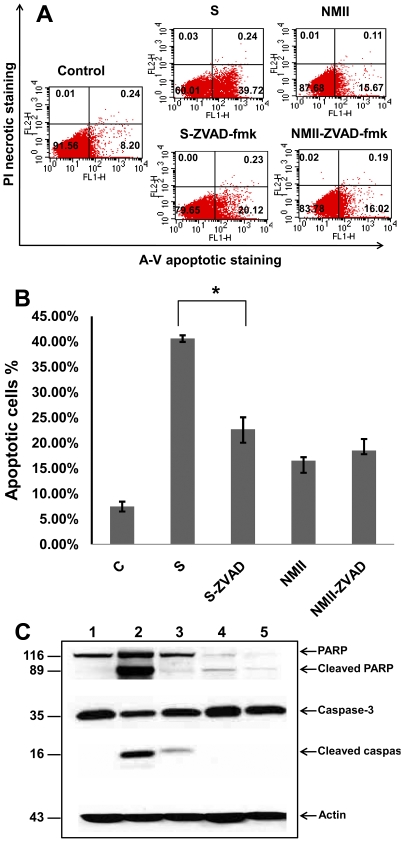
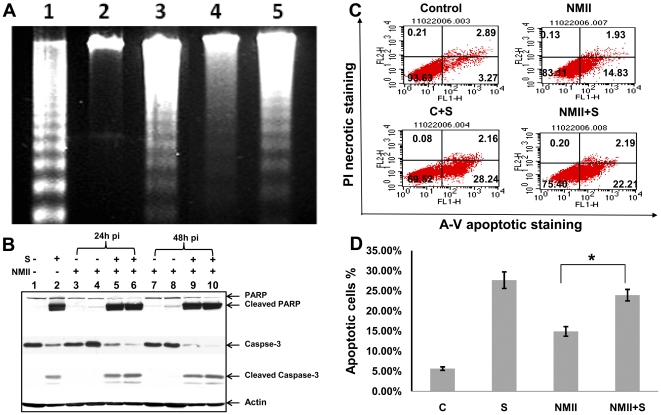
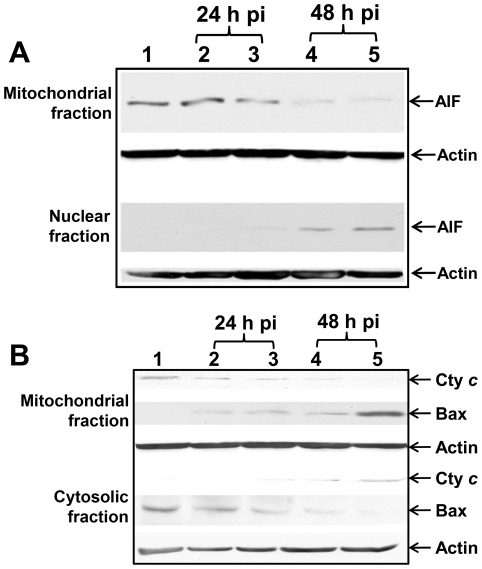
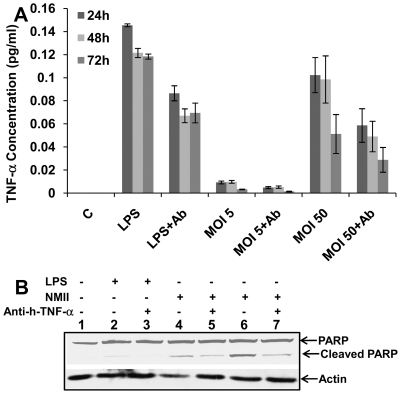
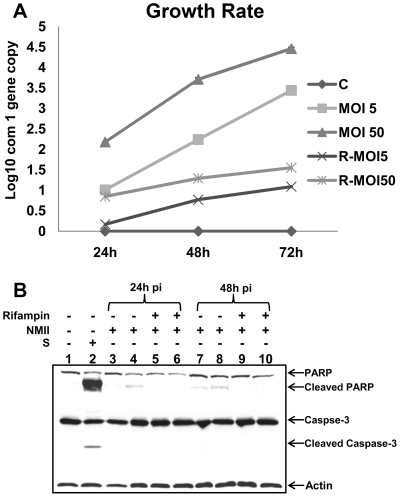
The ability of Coxiella burnetii to modulate host cell death may be a critical factor in disease development. In this study, human monocytic THP-1 cells were used to examine the ability of C. burnetii Nine Mile phase II (NMII) to modulate apoptotic signaling. Typical apoptotic cell morphological changes and DNA fragmentation were detected in NMII infected cells at an early stage of infection. FACS analysis using Annexin-V-PI double staining showed the induction of a significant number of apoptotic cells at an early stage of NMII infection. Double staining of apoptotic cell DNA and intracellular C. burnetii indicates that NMII infected cells undergoing apoptosis. Interestingly, caspase-3 was not cleaved in NMII infected cells and the caspase-inhibitor Z-VAD-fmk did not prevent NMII induced apoptosis. Surprisingly, the caspase-3 downstream substrate PARP was cleaved in NMII infected cells. These results suggest that NMII induces apoptosis during an early stage of infection through a caspase-independent pathway in THP-1 cells. In addition, NMII-infected monocytes were unable to prevent exogenous staurosporine-induced apoptotic death. Western blot analysis indicated that NMII infection induced the translocation of AIF from mitochondria into the nucleus. Cytochrome c release and cytosol-to-mitochondrial translocation of the pore-forming protein Bax in NMII infected cells occurred at 24 h post infection. These data suggest that NMII infection induced caspase-independent apoptosis through a mechanism involving cytochrome c release, cytosol-to-mitochondrial translocation of Bax and nuclear translocation of AIF in THP-1 monocytes. Furthermore, NMII infection increased TNF-α production and neutralization of TNF-α in NMII infected cells partially blocked PARP cleavage, suggesting TNF-α may play a role in the upstream signaling involved in NMII induced apoptosis. Antibiotic inhibition of C. burnetii RNA synthesis blocked NMII infection-induced PARP activation. These results suggest that both intracellular C. burnetii replication and secreted TNF-α contribute to NMII infection-triggered apoptosis during an early stage of infection.
Conflict of interest statement
Figures
Similar articles
-
Coxiella burnetii Inhibits Neutrophil Apoptosis by Exploiting Survival Pathways and Antiapoptotic Protein Mcl-1.Infect Immun. 2018 Mar 22;86(4):e00504-17. doi: 10.1128/IAI.00504-17. Print 2018 Apr. Infect Immun. 2018. PMID: 29311244 Free PMC article.
-
Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages.Infect Immun. 2007 Sep;75(9):4263-71. doi: 10.1128/IAI.00594-07. Epub 2007 Jul 2. Infect Immun. 2007. PMID: 17606599 Free PMC article.
-
Cinnamaldehyde-induced apoptosis in human hepatoma PLC/PRF/5 cells involves the mitochondrial death pathway and is sensitive to inhibition by cyclosporin A and z-VAD-fmk.Anticancer Agents Med Chem. 2013 Dec;13(10):1565-74. doi: 10.2174/18715206113139990144. Anticancer Agents Med Chem. 2013. PMID: 23438824
-
"Hairiness" is a Facsimile of Reorganized Cytoskeletons: A Cytopathic Effect of Coxiella burnetii.Yonsei Med J. 2019 Oct;60(10):890-897. doi: 10.3349/ymj.2019.60.10.890. Yonsei Med J. 2019. PMID: 31538423 Free PMC article. Review.
-
NAD+ depletion or PAR polymer formation: which plays the role of executioner in ischaemic cell death?Acta Physiol (Oxf). 2011 Sep;203(1):225-34. doi: 10.1111/j.1748-1716.2010.02229.x. Epub 2011 Jan 19. Acta Physiol (Oxf). 2011. PMID: 21091637 Free PMC article. Review.
Cited by
-
Tripping on acid: trans-kingdom perspectives on biological acids in immunity and pathogenesis.PLoS Pathog. 2013;9(7):e1003402. doi: 10.1371/journal.ppat.1003402. Epub 2013 Jul 18. PLoS Pathog. 2013. PMID: 23874196 Free PMC article. Review. No abstract available.
-
Primary Role for Toll-Like Receptor-Driven Tumor Necrosis Factor Rather than Cytosolic Immune Detection in Restricting Coxiella burnetii Phase II Replication within Mouse Macrophages.Infect Immun. 2016 Mar 24;84(4):998-1015. doi: 10.1128/IAI.01536-15. Print 2016 Apr. Infect Immun. 2016. PMID: 26787725 Free PMC article.
-
STING dependent BAX-IRF3 signaling results in apoptosis during late-stage Coxiella burnetii infection.Cell Death Dis. 2024 Mar 8;15(3):195. doi: 10.1038/s41419-024-06573-1. Cell Death Dis. 2024. PMID: 38459007 Free PMC article.
-
Coxiella burnetii Inhibits Neutrophil Apoptosis by Exploiting Survival Pathways and Antiapoptotic Protein Mcl-1.Infect Immun. 2018 Mar 22;86(4):e00504-17. doi: 10.1128/IAI.00504-17. Print 2018 Apr. Infect Immun. 2018. PMID: 29311244 Free PMC article.
-
Recent Advances on the Innate Immune Response to Coxiella burnetii.Front Cell Infect Microbiol. 2021 Nov 2;11:754455. doi: 10.3389/fcimb.2021.754455. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 34796128 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
