

Linking mitochondrial bioenergetics to insulin resistance via redox biology
- PMID: 22305519
- PMCID: PMC3313496
- DOI: 10.1016/j.tem.2011.12.008
Linking mitochondrial bioenergetics to insulin resistance via redox biology
Abstract
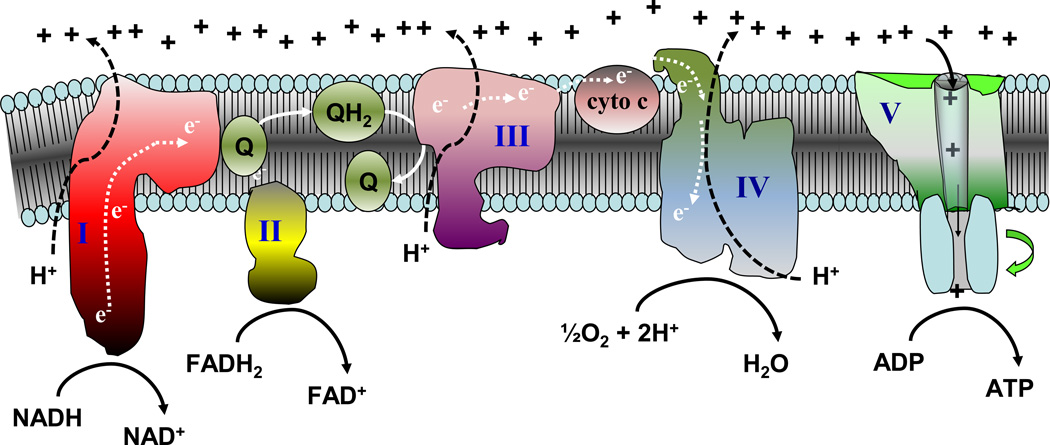
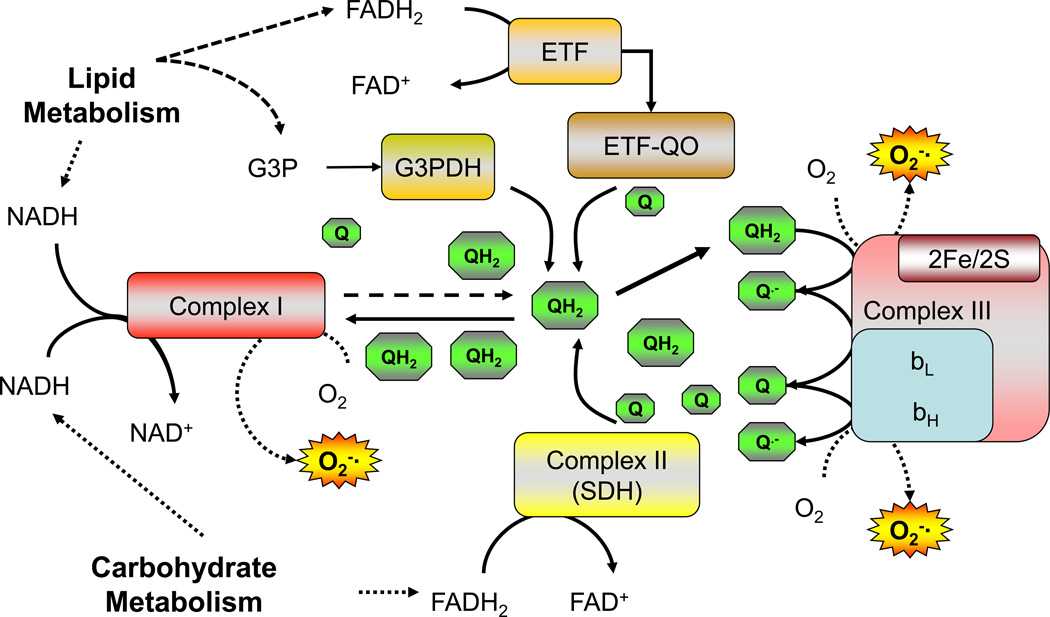
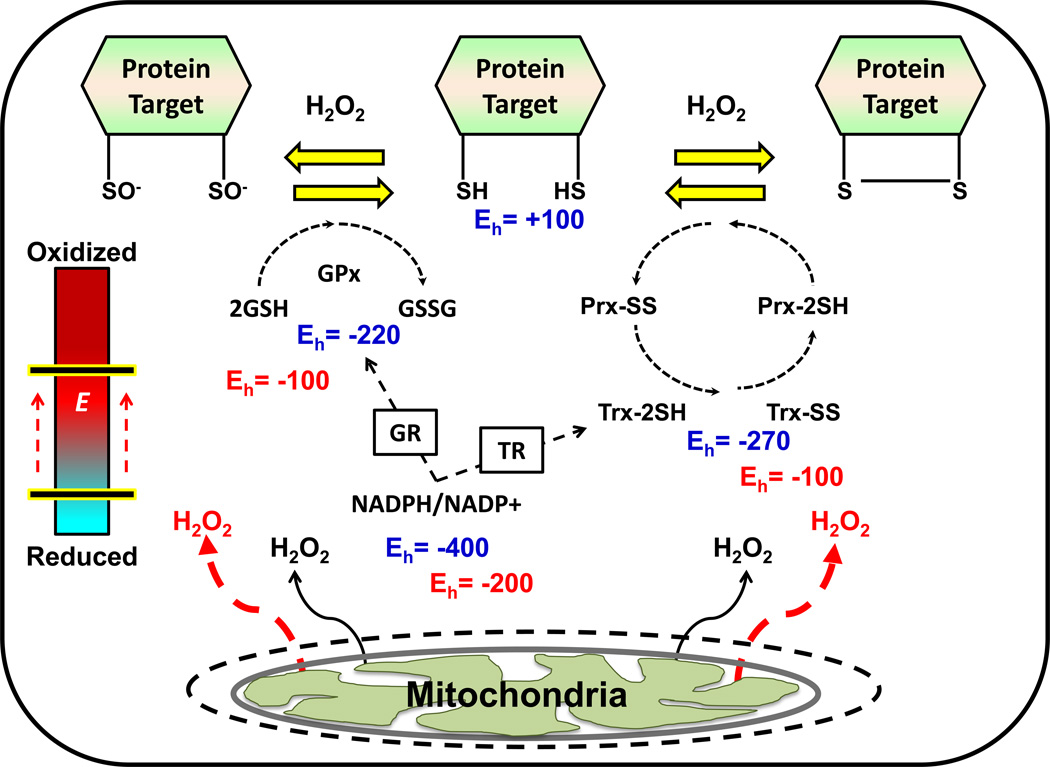
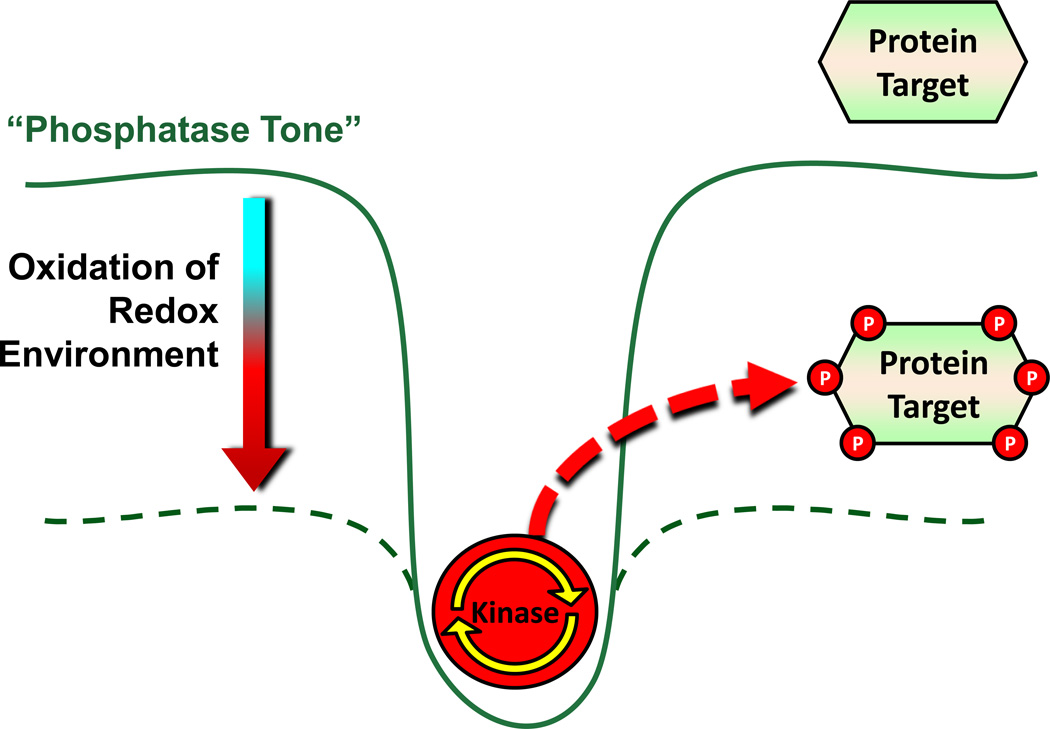
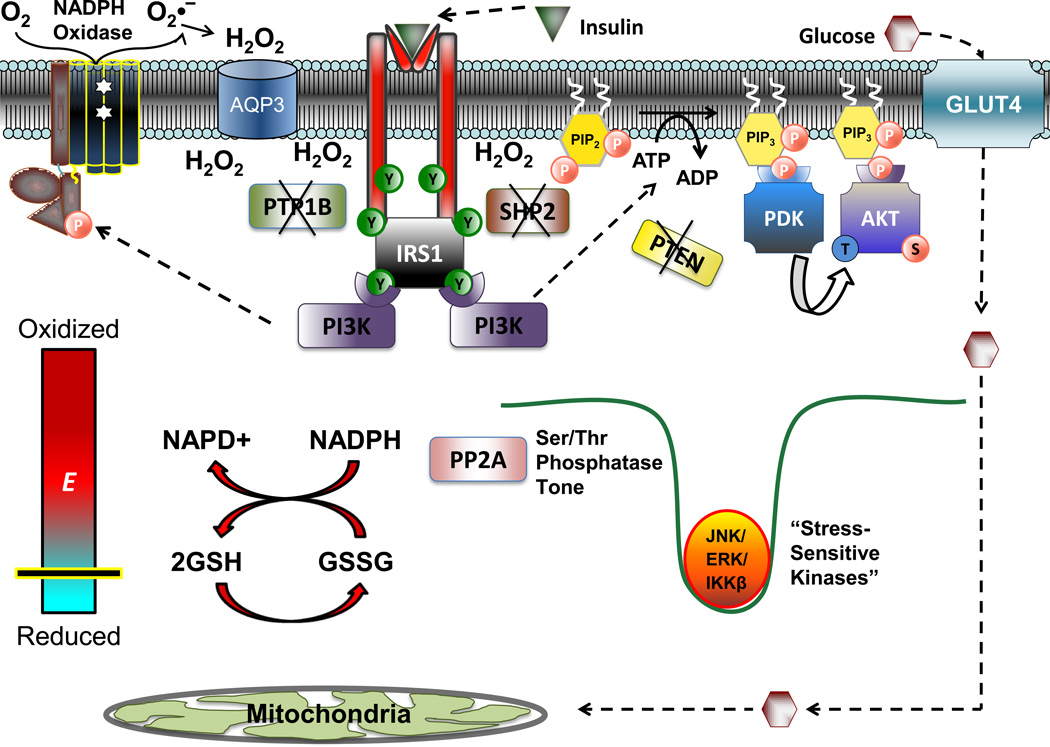
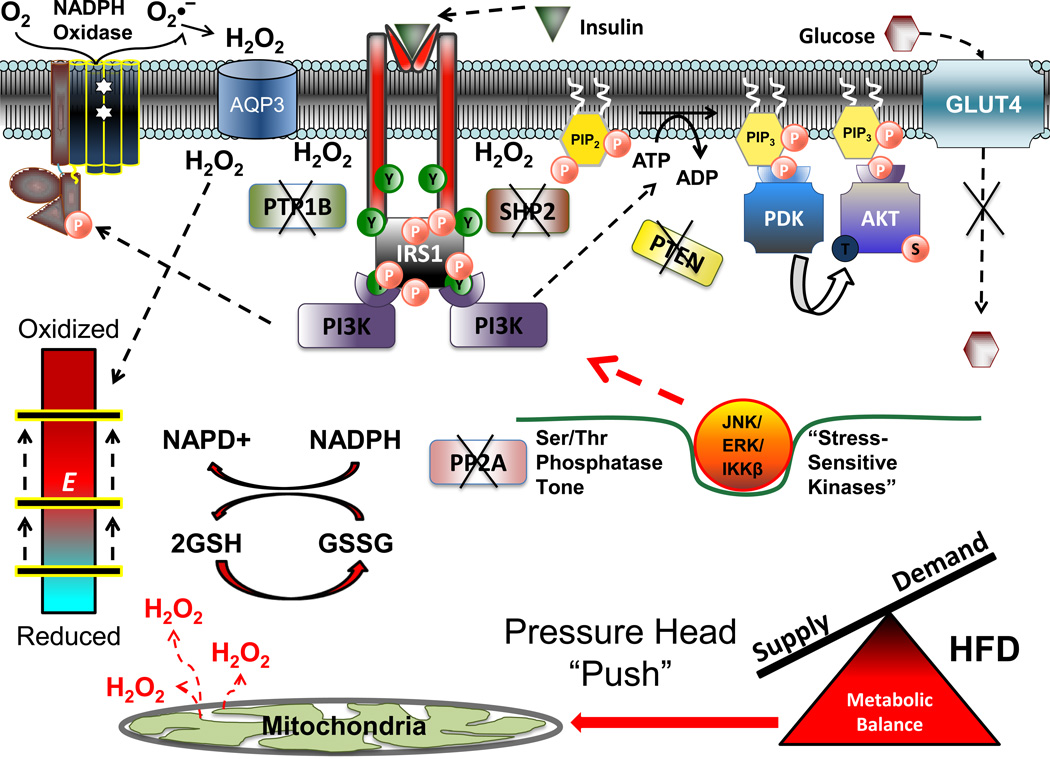
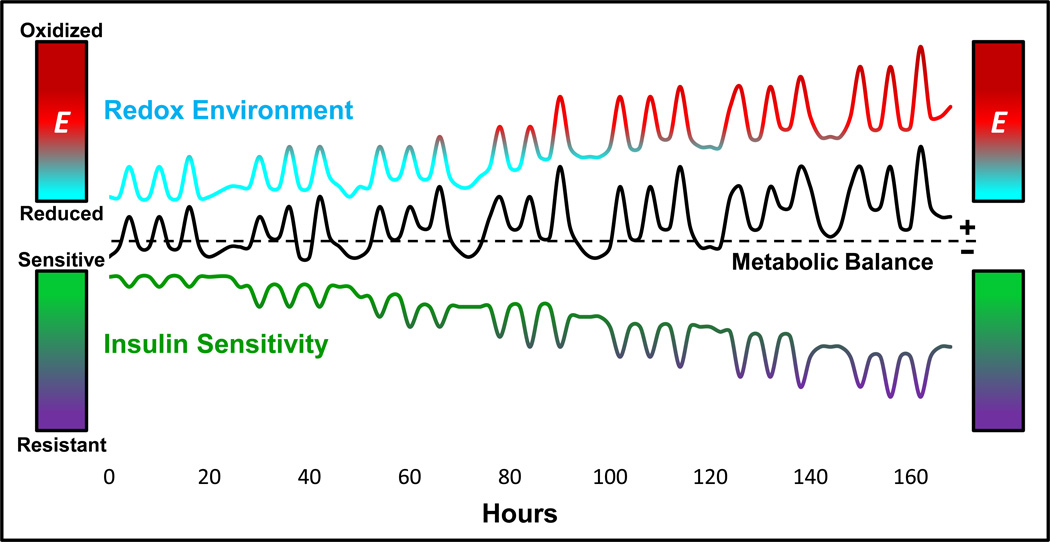
Chronic overnutrition and physical inactivity are major risk factors for insulin resistance and type 2 diabetes. Recent research indicates that overnutrition generates an increase in hydrogen peroxide (H(2)O(2)) emission from mitochondria, serving as a release valve to relieve the reducing pressure created by fuel overload, as well as a primary signal that ultimately decreases insulin sensitivity. H(2)O(2) is a major input to cellular redox circuits that link to cysteine residues throughout the entire proteome to regulate cell function. Here we review the principles of mitochondrial bioenergetics and redox systems biology and offer new insight into how H(2)O(2) emission may be linked via redox biology to the etiology of insulin resistance.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. - PubMed
-
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. - PubMed
-
- Korshunov SS, et al. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. - PubMed
-
- Liu SS. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci Rep. 1997;17:259–272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
