

RAD51 haploinsufficiency causes congenital mirror movements in humans
- PMID: 22305526
- PMCID: PMC3276668
- DOI: 10.1016/j.ajhg.2011.12.002
RAD51 haploinsufficiency causes congenital mirror movements in humans
Abstract
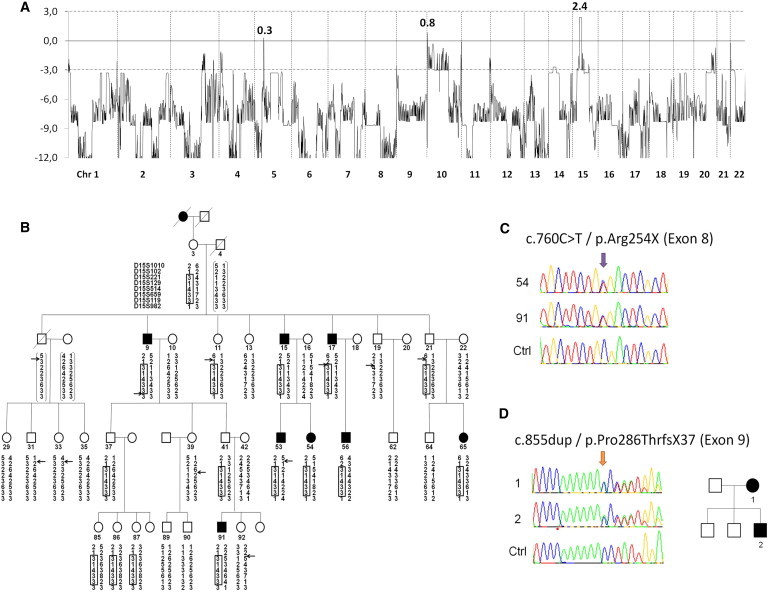
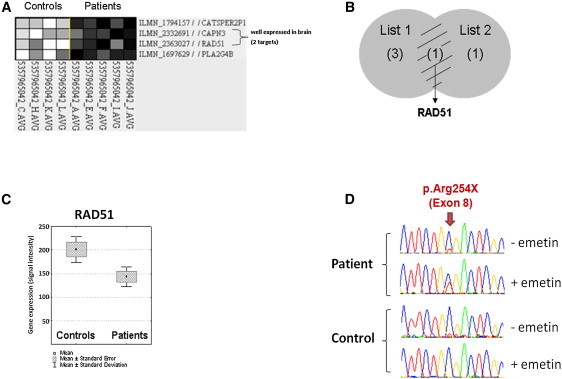
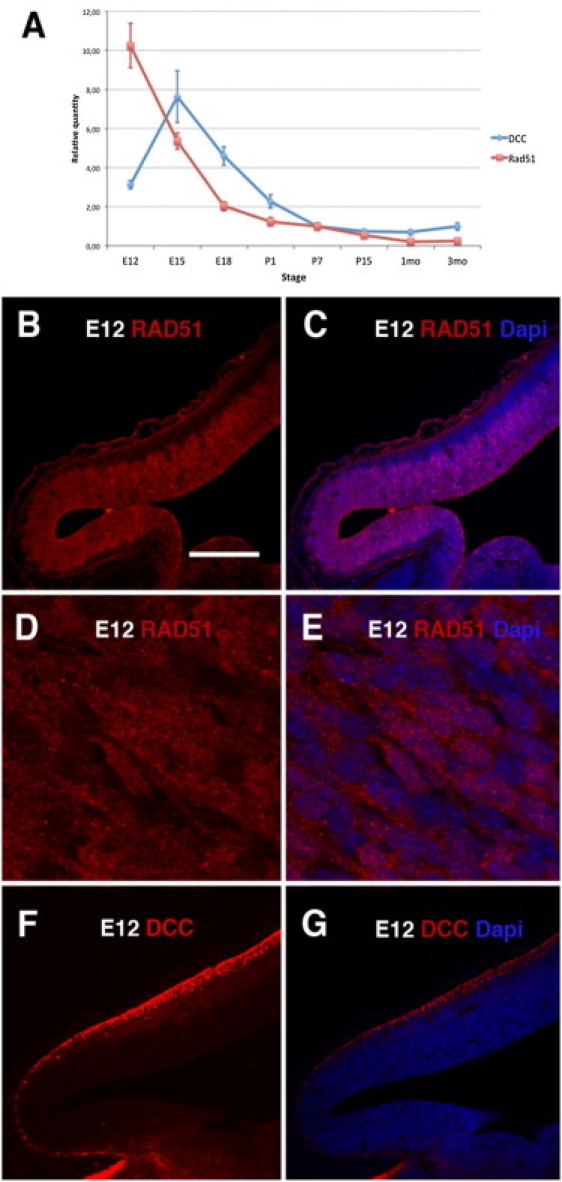
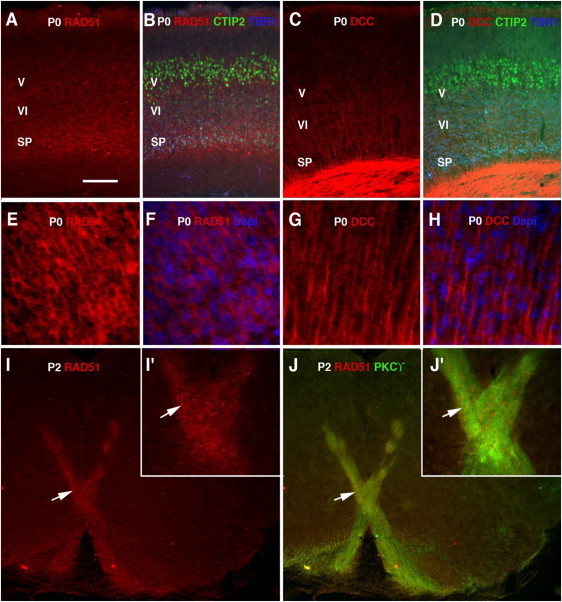
Congenital mirror movements (CMM) are characterized by involuntary movements of one side of the body that mirror intentional movements on the opposite side. CMM reflect dysfunctions and structural abnormalities of the motor network and are mainly inherited in an autosomal-dominant fashion. Recently, heterozygous mutations in DCC, the gene encoding the receptor for netrin 1 and involved in the guidance of developing axons toward the midline, have been identified but CMM are genetically heterogeneous. By combining genome-wide linkage analysis and exome sequencing, we identified heterozygous mutations introducing premature termination codons in RAD51 in two families with CMM. RAD51 mRNA was significantly downregulated in individuals with CMM resulting from the degradation of the mutated mRNA by nonsense-mediated decay. RAD51 was specifically present in the developing mouse cortex and, more particularly, in a subpopulation of corticospinal axons at the pyramidal decussation. The identification of mutations in RAD51, known for its key role in the repair of DNA double-strand breaks through homologous recombination, in individuals with CMM reveals a totally unexpected role of RAD51 in neurodevelopment. These findings open a new field of investigation for researchers attempting to unravel the molecular pathways underlying bimanual motor control in humans.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Bonnet C., Roubertie A., Doummar D., Bahi-Buisson N., Cochen de Cock V., Roze E. Developmental and benign movement disorders in childhood. Mov. Disord. 2010;25:1317–1334. - PubMed
-
- Galléa C., Popa T., Billot S., Méneret A., Depienne C., Roze E. Congenital mirror movements: a clue to understanding bimanual motor control. J. Neurol. 2011;258:1911–1919. - PubMed
-
- Srour M., Rivière J.B., Pham J.M., Dubé M.P., Girard S., Morin S., Dion P.A., Asselin G., Rochefort D., Hince P. Mutations in DCC cause congenital mirror movements. Science. 2010;328:592. - PubMed
-
- Depienne C., Cincotta M., Billot S., Bouteiller D., Groppa S., Brochard V., Flamand C., Hubsch C., Meunier S., Giovannelli F. A novel DCC mutation and genetic heterogeneity in congenital mirror movements. Neurology. 2011;76:260–264. - PubMed
-
- Djarmati-Westenberger A., Brüggemann N., Espay A.J., Bhatia K.P., Klein C. A novel DCC mutation and genetic heterogeneity in congenital mirror movements. Neurology. 2011;77:1580. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
