

Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs
- PMID: 22305566
- PMCID: PMC3682187
- DOI: 10.1016/j.stem.2011.12.017
Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs
Abstract
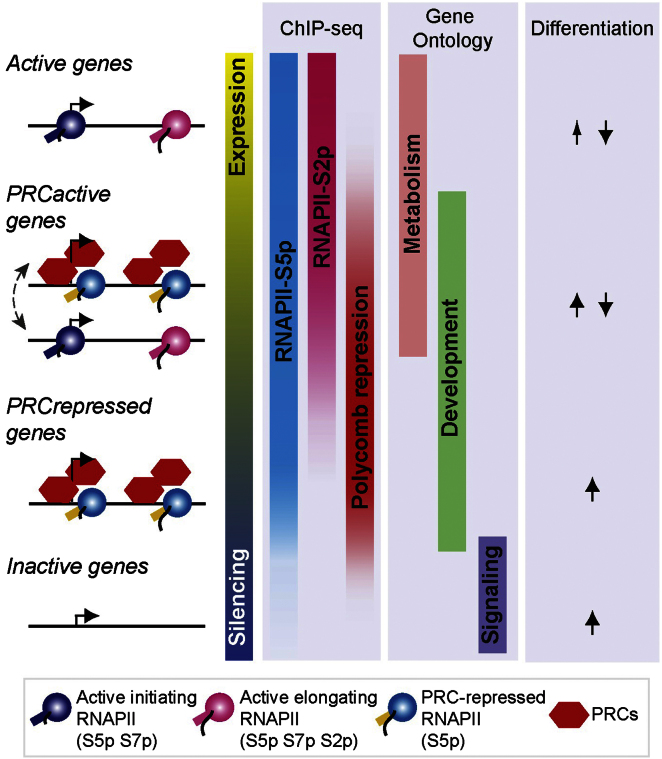
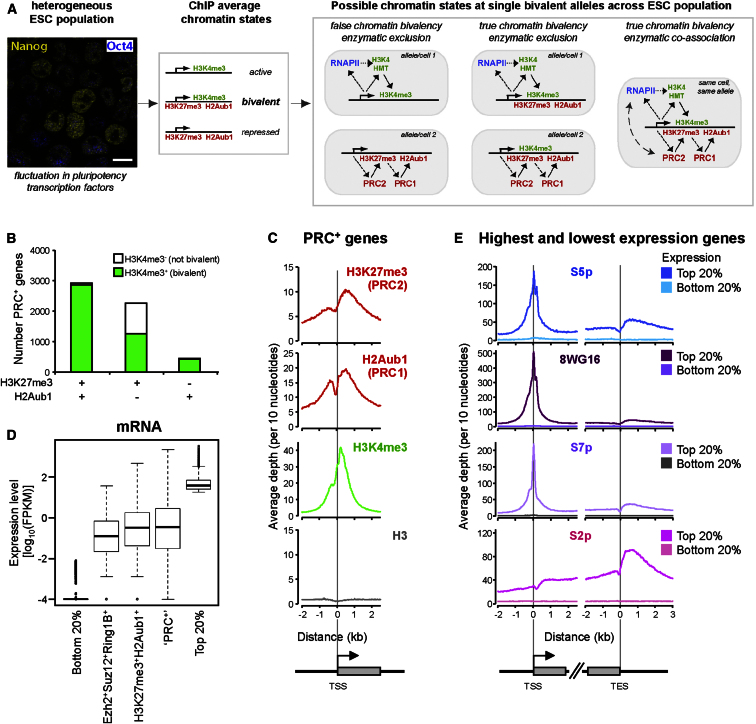
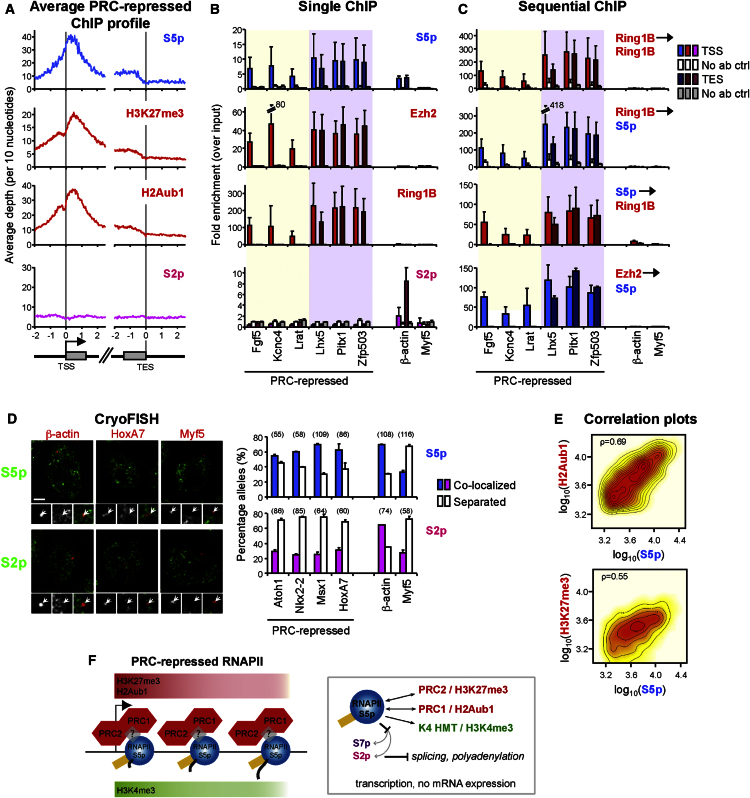
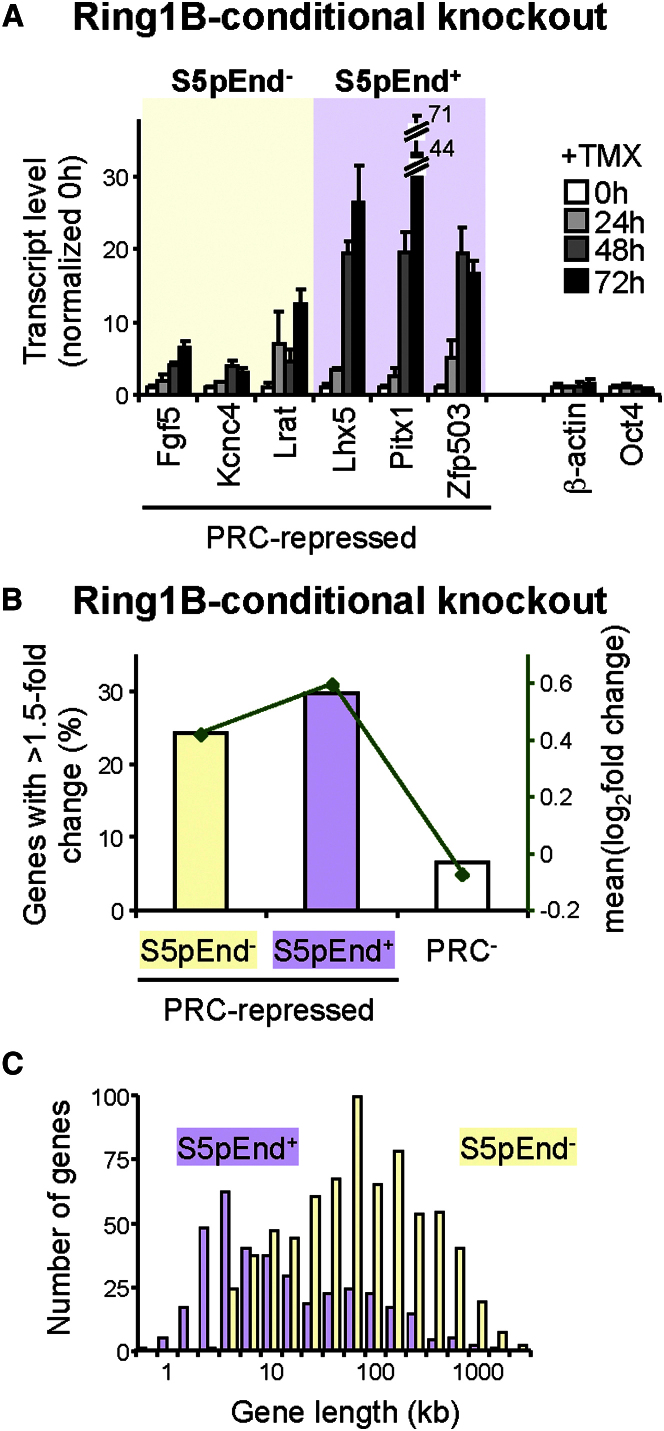
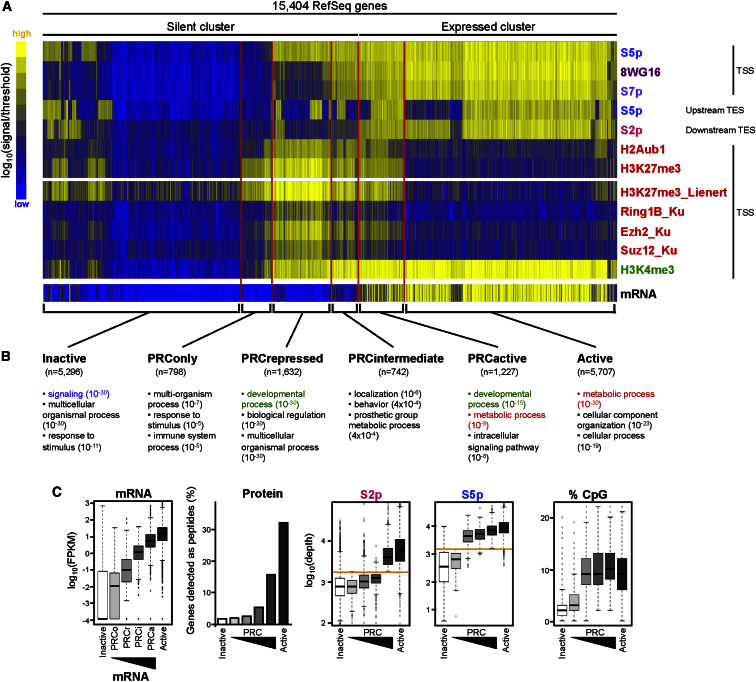
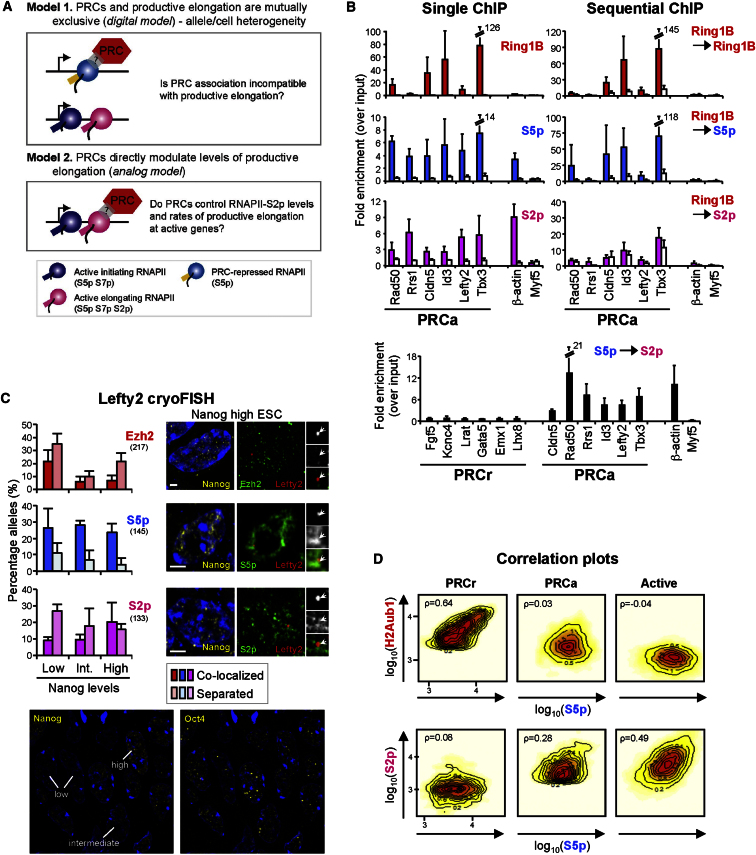
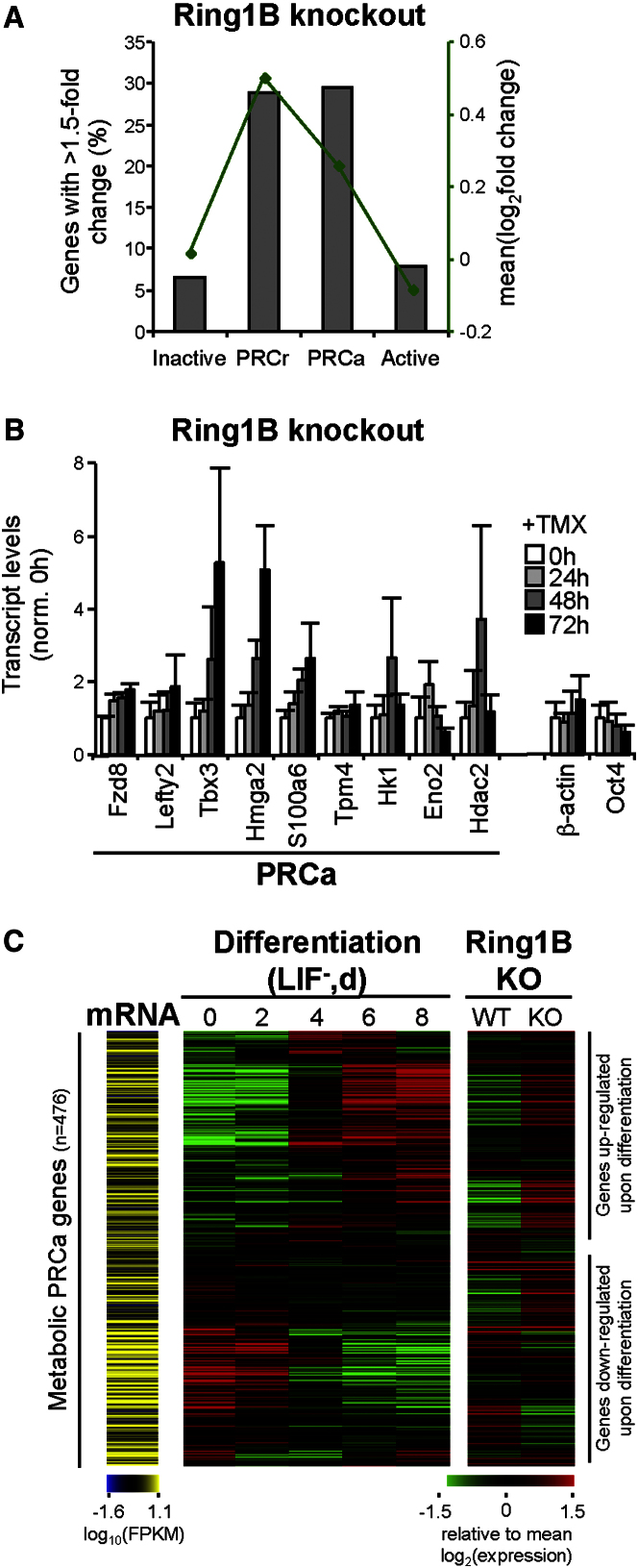
Polycomb repressor complexes (PRCs) are important chromatin modifiers fundamentally implicated in pluripotency and cancer. Polycomb silencing in embryonic stem cells (ESCs) can be accompanied by active chromatin and primed RNA polymerase II (RNAPII), but the relationship between PRCs and RNAPII remains unclear genome-wide. We mapped PRC repression markers and four RNAPII states in ESCs using ChIP-seq, and found that PRC targets exhibit a range of RNAPII variants. First, developmental PRC targets are bound by unproductive RNAPII (S5p(+)S7p(-)S2p(-)) genome-wide. Sequential ChIP, Ring1B depletion, and genome-wide correlations show that PRCs and RNAPII-S5p physically bind to the same chromatin and functionally synergize. Second, we identify a cohort of genes marked by PRC and elongating RNAPII (S5p(+)S7p(+)S2p(+)); they produce mRNA and protein, and their expression increases upon PRC1 knockdown. We show that this group of PRC targets switches between active and PRC-repressed states within the ESC population, and that many have roles in metabolism.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Alexa A., Rahnenführer J., Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
-
- Azuara V., Perry P., Sauer S., Spivakov M., Jørgensen H.F., John R.M., Gouti M., Casanova M., Warnes G., Merkenschlager M., Fisher A.G. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006;8:532–538. - PubMed
-
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
