

Computational redesign of a mononuclear zinc metalloenzyme for organophosphate hydrolysis
- PMID: 22306579
- PMCID: PMC3957331
- DOI: 10.1038/nchembio.777
Computational redesign of a mononuclear zinc metalloenzyme for organophosphate hydrolysis
Abstract
The ability to redesign enzymes to catalyze noncognate chemical transformations would have wide-ranging applications. We developed a computational method for repurposing the reactivity of metalloenzyme active site functional groups to catalyze new reactions. Using this method, we engineered a zinc-containing mouse adenosine deaminase to catalyze the hydrolysis of a model organophosphate with a catalytic efficiency (k(cat)/K(m)) of ~10(4) M(-1) s(-1) after directed evolution. In the high-resolution crystal structure of the enzyme, all but one of the designed residues adopt the designed conformation. The designed enzyme efficiently catalyzes the hydrolysis of the R(P) isomer of a coumarinyl analog of the nerve agent cyclosarin, and it shows marked substrate selectivity for coumarinyl leaving groups. Computational redesign of native enzyme active sites complements directed evolution methods and offers a general approach for exploring their untapped catalytic potential for new reactivities.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
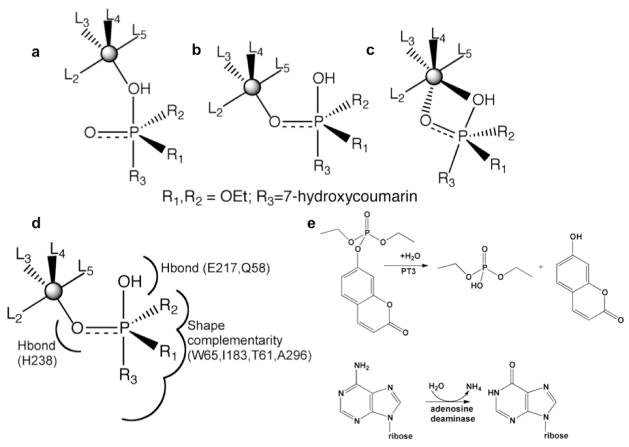
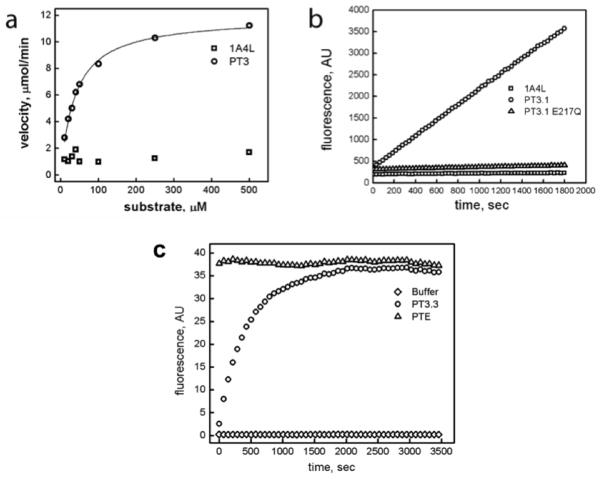
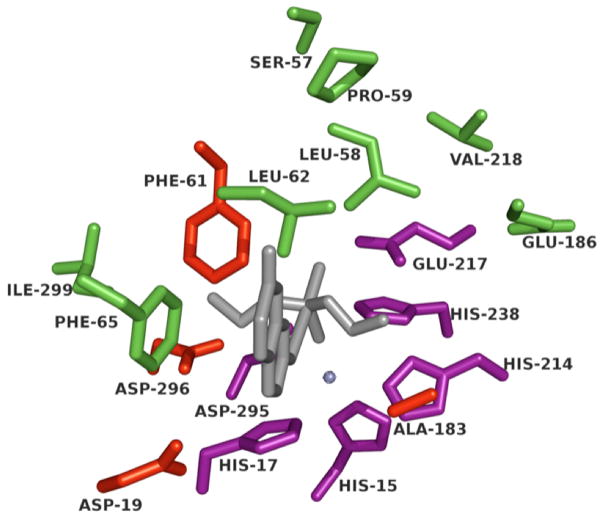
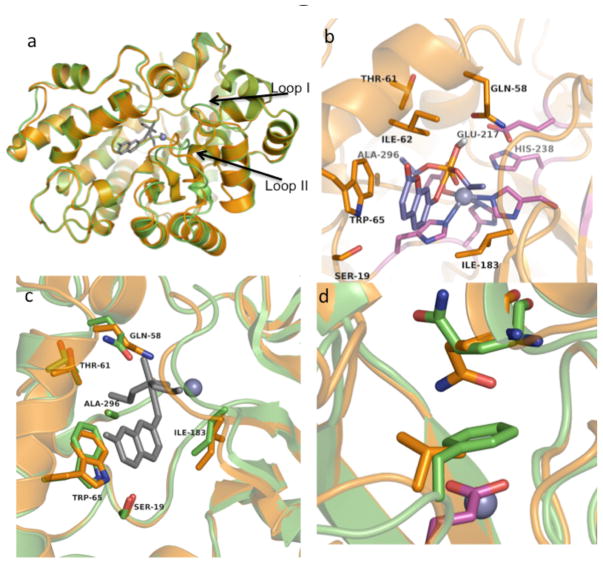
Comment in
-
Protein design: A metalloenzyme reloaded.Nat Chem Biol. 2012 Feb 15;8(3):224-5. doi: 10.1038/nchembio.800. Nat Chem Biol. 2012. PMID: 22337091 No abstract available.
References
-
- Toscano MD, Woycechowsky KJ, Hilvert D. Minimalist active-site redesign: Teaching old enzymes new tricks. Angewandte Chemie-International Edition. 2007;46:3212–3236. - PubMed
-
- Khersonsky O, Tawfik DS. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annual Review of Biochemistry. 2010;79:471–505. - PubMed
-
- Terao Y, Miyamoto K, Ohta H. Introduction of single mutation changes arylmalonate decarboxylase to racemase. Chemical Communications. 2006:3600–3602. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
