

Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice
- PMID: 22307032
- PMCID: PMC3482511
- DOI: 10.1053/j.gastro.2012.01.032
Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice
Abstract
Background & aims: Obesity-related insulin resistance contributes to cardiovascular disease. Cannabinoid receptor-1 (CB(1)) blockade improves insulin sensitivity in obese animals and people, suggesting endocannabinoid involvement. We explored the role of hepatic CB(1) in insulin resistance and inhibition of insulin signaling pathways.
Methods: Wild-type mice and mice with disruption of CB(1) (CB(1)(-/-) mice) or with hepatocyte-specific deletion or transgenic overexpression of CB(1) were maintained on regular chow or a high-fat diet (HFD) to induce obesity and insulin resistance. Hyperinsulinemic-euglycemic clamp analysis was used to analyze the role of the liver and hepatic CB(1) in HFD-induced insulin resistance. The cellular mechanisms of insulin resistance were analyzed in mouse and human isolated hepatocytes using small interfering or short hairpin RNAs and lentiviral knockdown of gene expression.
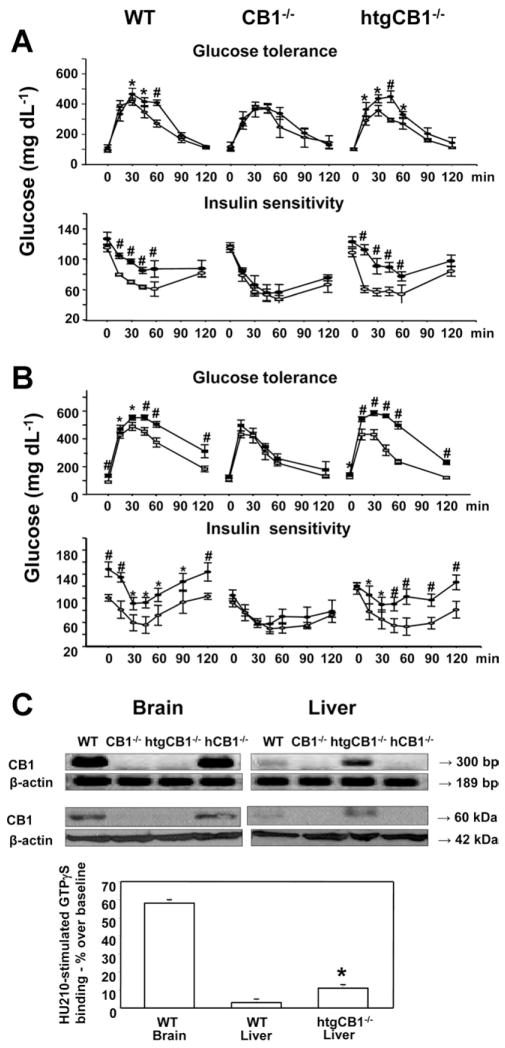
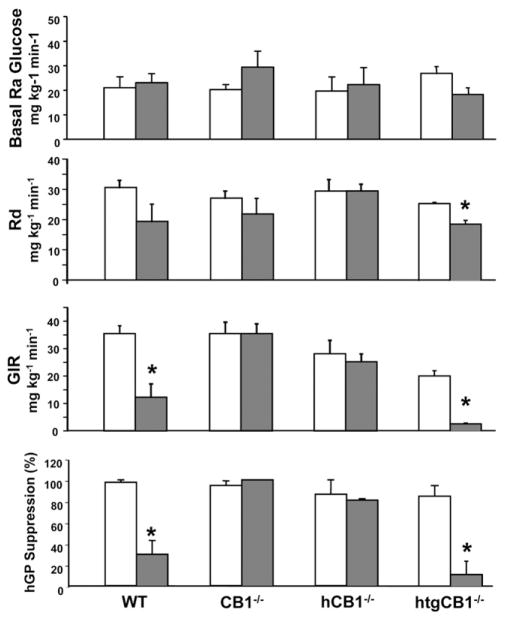
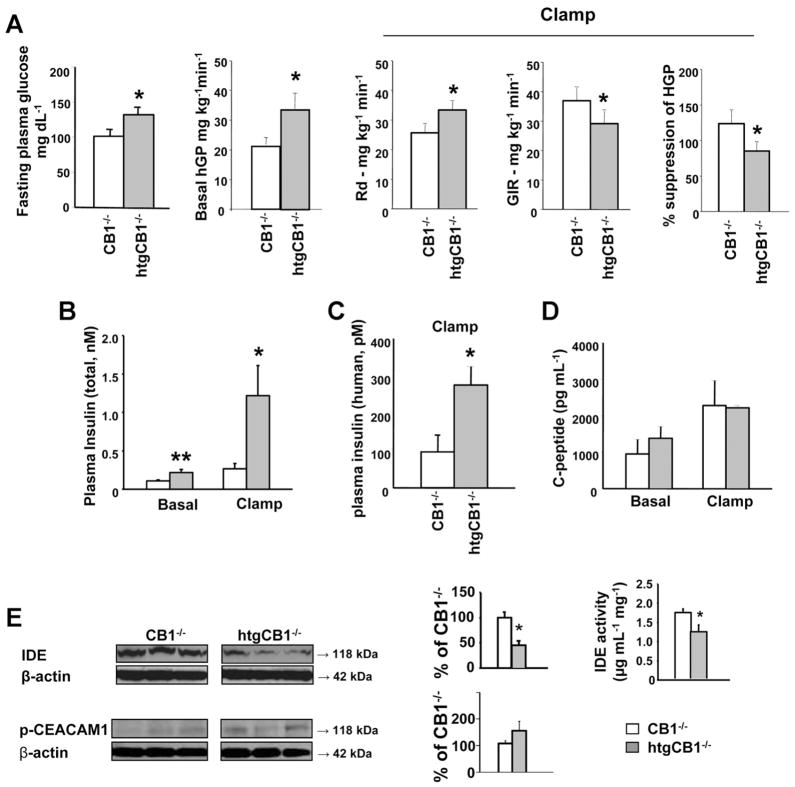
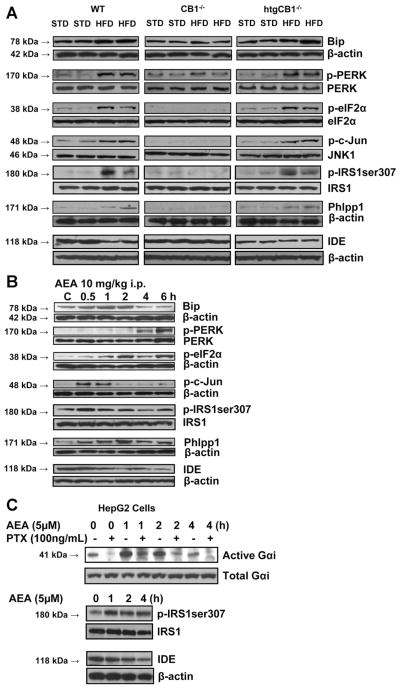
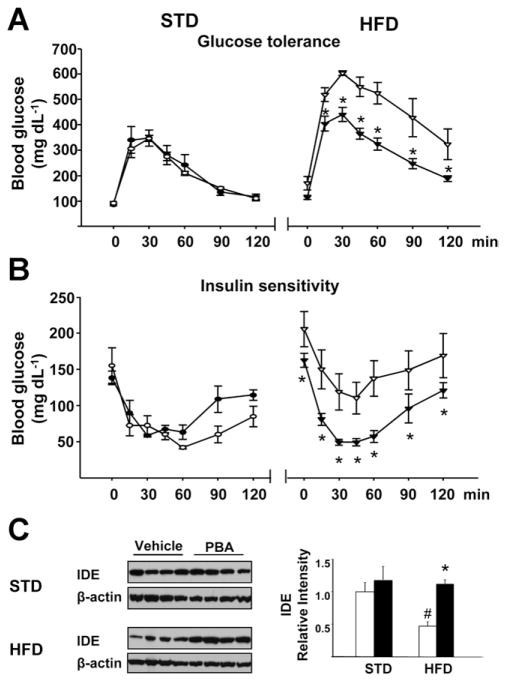
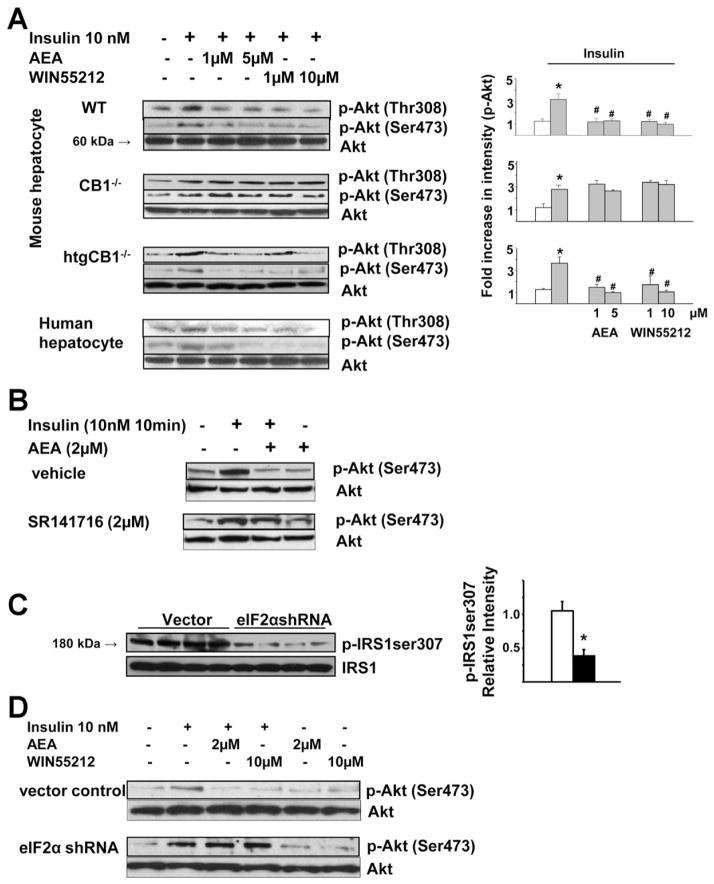
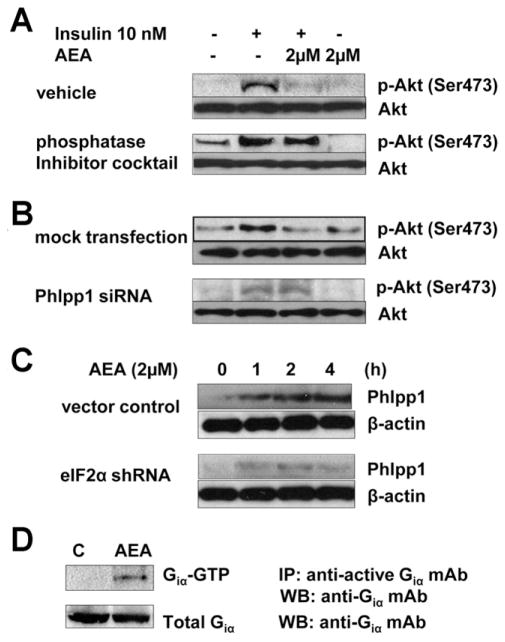
Results: The HFD induced hepatic insulin resistance in wild-type mice, but not in CB(1)(-/-) mice or mice with hepatocyte-specific deletion of CB(1). CB(1)(-/-) mice that overexpressed CB(1) specifically in hepatocytes became hyperinsulinemic as a result of reduced insulin clearance due to down-regulation of the insulin-degrading enzyme. However, they had increased hepatic glucose production due to increased glycogenolysis, indicating hepatic insulin resistance; this was further increased by the HFD. In mice with hepatocytes that express CB(1), the HFD or CB(1) activation induced the endoplasmic reticulum stress response via activation of the Bip-PERK-eIF2α protein translation pathway. In hepatocytes isolated from human or mouse liver, CB(1) activation caused endoplasmic reticulum stress-dependent suppression of insulin-induced phosphorylation of akt-2 via phosphorylation of IRS1 at serine-307 and by inducing the expression of the serine and threonine phosphatase Phlpp1. Expression of CB(1) was up-regulated in samples from patients with nonalcoholic fatty liver disease.
Conclusions: Endocannabinoids contribute to diet-induced insulin resistance in mice via hepatic CB(1)-mediated inhibition of insulin signaling and clearance.
Copyright © 2012 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
The authors disclose no conflicts.
Figures
Comment in
-
"De-liver-ance" from CB(1): a way to counteract insulin resistance?Gastroenterology. 2012 May;142(5):1063-6. doi: 10.1053/j.gastro.2012.03.011. Epub 2012 Mar 27. Gastroenterology. 2012. PMID: 22465721 No abstract available.
References
-
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
